Синдром олбрайта. Синдром Олбрайта: причины, симптомы, лечение. Данное заболевание в основном возникает у девочек
Синдром Мак-Кьюна-Олбрайта является гетерогенным заболеванием, которое независимо друг от друга описали американский педиатр D. J. McCune и эндокринолог F. Albright в 1936-1937 гг. Заболевание редкое, врожденное, но не наследственное, сопровождается метаболическими нарушениями, признаками преждевременного полового развития и деформаций скелета. Первые клинические проявления отмечаются в раннем детстве или подростковом возрасте в виде нарушений развития, полиоссальной фиброзной дисплазии костей, кожной пигментации по типу «кофе с молоком» и эндокринных аномалий, включая , синдром Кушинга , .
Приблизительно в 10 раз чаще диагностируют синдром Олбрайта Мак-Кьюна у девочек, чем у мальчиков. Википедия отмечает, что частота встречаемости в человеческой популяции — 1 на 100 тыс. или на миллион особ.
Патогенез
В результате постзиготной мутации гена GNAS1 , участвующего в клеточном сигнализировании белков GS-α , обеспечивающих сопряжение рецепторов лютеинизирующих и фоликолостимулирующих гормонов с молекулами аденилатциклазы в цитологических структурах яичников. Постоянное влияние мутантного белка активизирует аденилатциклазу и повышение внутриклеточного цАМФ , а также стимулирует усиление секреции стероидных женских половых гормонов фолликулярными эстрогенпродуцирующими кистами, вызывающими преждевременное половое развитие даже при отсутствии . Такая же картина наблюдается и «автономное производство» наблюдается с гормонами щитовидной железы, и . Прогрессирование патологии вызвано образованием клеток, уже содержащих мутантные белки.
Кроме того, рост уровня цАМФ приводит к увеличению продукции и образованию лентигиозных пятен, очаговой гиперпигментации на фоне нормальных значений и меланотропина .
Фиброзно-кистозная дисплазия в результате усиленной кальцинации под действием эстрогенов осуществляется путем замещения нормальных костных структур и образованных кальцинатов фиброзными массами, при этом нарушается равновесие процессов резорбции и регенерации костей, пролиферации мезенхимальных стромальных клеток. Наблюдается формирование очагов деструкций и псевдоопухолей.
Классификация
Болезнь Олбрайта бывает разной, в зависимости от тяжести течения различают:
- скрытое заболевание – без каких-либо признаков костной или эндокринной патологии, а также без симптоматики необычной пигментации;
- неполные формы синдрома , например, в процесс патогенеза вовлечения костная ткань, возможны эндокринные изменения и псевдогипопаратиреоз , но нет кожных проявлений;
- псевдогипопаратиреоз или наследственная остеодистрофия Олбрайта — заболевание, которое также поражает костные ткани, но механизм и этиология отличаются — патология вызвана нарушением резистентности к паратгормону , метаболизма кальция и фосфора; псевдогипопаратиреоз также проявляется в детском возрасте и выражается в виде низкого роста, круглого лица, умственной отсталости, гипогонадизма , и пр.

Причины
Синдром Олбрайта Маккьюна возникает в результате генетических нарушений на ранних этапах эмбриогенеза. До сих пор не установлен тип наследования, все описанные случаи были спорадические.
Симптомы
Клиническая картина заболевания проявляется в виде целого симпатокомплекса, состоящего из:
- преждевременного гонадотропиннезависимого полового развития, развивающегося позже и медленнее нежели при других формах ранней половозрелости;
- пятнистой пигментации кожи – лентиго , пятна с четкими границами светло-коричневого цвета обычно есть от рождения на груди, спине пояснице, бедрах и в местах костных деформаций;
- эндокринных расстройствах, вызывающих , необъяснимую утрату веса, глазные симптомы гипертиреоидизма ;
- полиоссальной фиброзной остеодисплазии , и , вовлекающей в процесс чаще длинные кости, нарушения проявляются в виде изуродованных черт лица, более вытянутом телосложении, искривленных и асимметричных, укороченных запястьях, что затрудняет движения во время ходьбы, вызывает хромоту, сильные боли и повышает вероятность патологических переломов.

Первые признаки преждевременной половой зрелости начинаются с маточных кровотечений, не имеющих цикличного характера и вызваны кратковременными повышениями концентраций эстрогенов и гонадотропных гормонов в кровяном русле. Такие колебания провоцируют образования кист в яичниках. У детей в возрасте 5-10 лет наблюдается ранний рост груди и гинекомастия . Её нежная этиология не приводит к формированию вторичных половых признаков — Синдром Мак-Кьюна-Олбрайта-Брайцева не вызывает раннего полового оволосения или дифференцировки фигуры. Истинное половое созревание возможно после окончания пубертатного периода.
Анализы и диагностика
При подозрении на синдром Мак Кьюна Олбрайт проводят:
- рентгенологические исследования, которые выявляют псевдокисты , сегментарное поражение любых отделов скелета – чаще нижних конечностей, в более редких случаях — верхнего плечевого пояса и черепа;
- УЗИ брюшной полости позволяет обнаружить ;
- лабораторные исследования для выявления повышенного уровня щелочной фосфатазы в кровотоке, сниженного количества гонадотропинов – в анализах мочи и повышенной экскреции лютеинизирующего гормона , 17-гидрокси и 17-окси кортикостероидов .
Лечение
Лечение назначают симптоматическое, оно имеет свои сложности в зависимости от индивидуальных особенностей и клинической картины. Аналоги гонадолиберина не оказывают терапевтического эффекта, так как процесс секреции эстрогенов спровоцирован не избытком гонадотропинов. Для купирования процесса гонадального стероидогенеза у особ мужского пола эффективным может оказываться и . При лечении медроксипрогестерона ацетатом возможно развитие гипокальциемии , поэтому, если процесс развития ранней половозрелости сопровождается тяжёлыми множественными поражениями костных тканей, то терапию необходимо назначать с осторожностью.
Для угнетения гиперэстрогенизиции и удлинения скелета могут быть назначены препараты . Чтобы противодействовать дисплазии костей следует вводить:
- бисфосфонаты ;
- ингибиторы антагонистов остеокластов.
Больным необходимо регулярно, каждые 3 мес. посещать эндокринолога для коррекции проблем с щитовидной и другими железами.
Доктора
Лекарства
- – несмотря на то, что более известен как противогрибковый препарат, в больших дозах он оказывается эффективен при кушиноподобных проявлениях. Суточная доза, разделенная на 2-3 приема, может достигать 1200 мг.
- Медроксипрогестерон – обладает андрогенным, противоопухолевым и анаболическим действием. Возможны различные пути введения в организм в дозах, индивидуально установленных лечащим врачом.
- Памидроновая кислота – препарат способен предотвратить остеолитическую деструкцию костей. Можно вводить в/в струйно, но не следует сочетать с лечением другими бисфосонатами.
- – бисфосфонат, способствующий формированию костей с нормальными гистологическим строением, их восстановлению и повышению минеральной плотности. Препарат следует принимать перорально внутрь – по 1 таблетке в неделю.
Процедуры и операции
- Овариэктомия – удаление яичников может быть рекомендовано в случаях чрезмерного прогрессирования гиперпубертатного развития.
- Хирургическая реконструкция деформации костей скелета и черепа.
Прогноз
Последние годы о синдроме МакКьюна Олбрайт и проблемах его лечения стали говорить все чаще, а особы, страдающие от этого недуга стали заявлять о себе и раскрывать особенности бытовой и социальной жизни в личных блогах и видео. Благодаря современным технологиям, не смотря на неизлечимость патологии, качество жизни таких людей значительно возросло, теперь они могут найти способ проявить себя и даже снимаются в кино, например, как уругвайский актер Маурисио Саравия.
Список источников
- McCune. D. J. Osteitis fibrosa cystica: the case of a nine year old girl who also exhibits precocious puberty, multiplepigmentation of the skin and hyperthyroidism. American Journal of Diseases of Children, Chicago, 1936; 52: 743–747.
- Мандель Б. Р. Основы современной генетики. Учебное пособие. М:Флинта, 2017. – 305 С.
- Кулаков В.И., Манухина И.Б., Савельева Г.М. Гинекология: нац. рук. М. : ГЭОТАР-Медиа, 2011.- 461 с.
Данная информация предназначена для специалистов в области здравоохранения и фармацевтики. Пациенты не должны использовать эту информацию в качестве медицинских советов или рекомендаций.
Случай синдрома Мак-Кьюна - Олбрайта - Брайцева
Врач травматолог-ортопед высшей категории Багриновская Ирина
Лейбовна
Московская детская городская поликлиника №110
Ключевые слова: гиперпигментация / девочки / дети / лентиго / мутация / патологический перелом / полиоссальная остеодисплазия / преждевременное половое развитие / синдром Мак-Кьюна- Олбрайта- Брайцева / фолликулярная киста.
Синдром Мак-Кьюна- Олбрайта- Брайцева (синдром МОБ, McCune–Albright–Braitsev syndrome) или синдром Олбрайта- Мак-Кьюна- Стернберга (Albright–McCune–Sternberg syndrome), как указано под кодом Q78.1 в 10-й Международной классификации болезней. Первое указание на данное заболевание принадлежит русскому и советскому хирургу В.Р. Брайцеву, который в 1907г. описал фиброзно-кистозную болезнь челюсти (displasia fibrosa polycistia ) у детей с пигментными пятнами. Однако название синдром получил лишь после публикаций американского педиатра D.J.McCune в 1936г. и американского эндокринолога F.Albright в соавторстве с патологом W.Sternberg в 1937г.
Клинические проявления синдрома:
– асимметричные гиперпигментированные пятна цвета кофе с молоком (лентиго), обычно на коже груди, спины, в области поясницы и бедер;
– эндокринопатии, чаще всего преждевременное половое развитие;
– полиоссальная фиброзно-кистозная остеодисплазия.
Синдром МОБ обусловлен соматической мутацией гена GNASI, расположенного на длинном плече 20-й хромосомы, на ранних стадиях эмбриогенеза, в связи с чем не передается по наследству. В результате образуются клоны мутантных клеток. Ген GNASI, в частности, кодирует α-субъединицу G-белка (Gαs), служащего посредником в превращении циклического аденозинмонофосфата (цАМФ), который регулирует гормональную систему человека.
При росте уровня цАМФ возрастает продукция меланина при нормальных значениях АКТГ и меланостимулирующего гормона, вследствие чего образуются лентигинозные пятна.
Так как Gαs служит медиатором в передаче сигнала от ЛГ и ФСГ, то мутантные клетки половых желез проявляют резко повышенную автономную реактивность, что приводит у девочек к возникновению фолликулярных эстрогенпродуцирующих кист и преждевременному половому развитию . Мутантные клетки распределяются в гонадах неравномерно. У девочек они могут локализоваться в одном или двух яичниках, обусловливая периодическое формирование односторонних либо двусторонних активных кист с их последующим регрессом .
Активная Gαs инициирует нарушение равновесия между резорбцией и регенерацией костей, пролиферацию незрелых стромальных клеток, что приводит к замещению нормальной костной структуры фиброзными массами и формированием очагов деструкции в виде кист .
Опосредуя эффекты паратиреоидного гормона (ПТГ), Gαs участвует в поддержании гомеостаза костной ткани и кальций-фосфорного обмена. ПТГ служит основным фактором, отвечающим за гомеостаз кальциевого обмена. Кальций вымывается из костей и откладывается в виде кальцинатов в подкожно-жировой клетчатке, мышцах, внутренних органах, эндотелии артериальных сосудов. Со временем костная ткань замещается фиброзной. Скелетные аномалии нарушают мобильность больных и становятся причиной сильной боли. Снижение витаминаD в крови приводит к нарушению процесса всасывания кальция в кишечнике. Гипокальциемия стимулирует еще большее выделение в кровь ПТГ и усиленное вымывание кальция из костей, что приводит к развитию выраженной остеомаляции .
В процессе роста организма и возрастающей весовой нагрузки патологический участок кости искривляется. У ряда пациентов бедренная кость деформируется и в результате патологических переломов приобретает форму пастушьего посоха. У некоторых больных сильная деформация костей приводит к инвалидизации .
При наличии мутантных клеток в других железах эндокринной системы, могут возникать многоузловой зоб, гипофосфатемический рахит, синдром Кушинга, акромегалия идр. При поражении кардиомиоцитов наблюдается тахикардия. Возможны поражения желудочно-кишечного тракта, такие как холестатический гепатит, гастроинтестинальные полипы, гастроинтестинальный рефлюкс .
Этому заболеванию подвержены оба пола, однако у девочек оно встречается в 2раза чаще, чем у мальчиков. Преждевременное половое развитие у девочек отмечается в 9раз чаще, чем у мальчиков .
Синдром МОБ- редкое (орфанное) заболевание. Частота встречаемости в мире варьирует от 1:100000 до 1:1000000 в общей популяции.
Течение и прогноз. Заболевание клинически неоднородное. Наряду со случаями классической триады признаков, существуют атипичные и неполные формы синдрома. Выраженность симптомов и тяжесть заболевания также сильно изменчивы. С возрастом патология костной ткани прогрессирует, однако при стертых формах прогрессирование замедляется с наступлением пубертата. Развитие только костной патологии по частоте превышает полный симптомокомплекс в 30-40 раз.
Клиническое наблюдение. Девочка В. 12лет поступила на прием к врачу травматологу-ортопеду Московской детской городской поликлиники №110 31.05.2017г.
Жалобы при поступлении на боли в ногах, усиливающиеся после длительной ходьбы или занятий бальными танцами. По словам пациентки, боли такой силы, что их невозможно терпеть, приходится останавливаться и садиться в любом месте. Считает себя больной в течение 3мес.
Анамнез семейный: родители- русские по национальности, не состоящие в кровном родстве. Случаев психических и неврологических заболеваний, задержки развития в родословной не было. Сибс, сестра 15лет, со слов матери здорова.
Анамнез жизни: ребенок от второй беременности, роды в срок, физиологические, масса при рождении 3500г, рост 51см, закричала сразу, оценка по шкале Апгар 8/9 баллов. Голову начала держать в 2мес, сидеть в 7мес, самостоятельно ходить в 1год, Грудное вскармливание до 5мес. Привита по возрасту. Инфекционные заболевания: ОРВИ, ветряная оспа.
Данные объективного исследования. Рост 158см, вес 40кг. Артериальное давление 120/75ммрт.ст., частота сердечных сокращений 78/мин. Общее состояние удовлетворительное. Функциональный статус: ходит, походка не нарушена. Телосложение правильное, нормостеническое. На правом и левом плече, на боковой поверхности туловища справа единичные мелкие светло-коричневые лентигинозные пятна с неровными краями диаметром до 20мм. Подкожно-жировая клетчатка развита умеренно, расположена равномерно. Видимые слизистые чистые, розовые, влажные. Периферические лимфоузлы единичные, мелкие, подвижные, безболезненные. Мышечная масса развита достаточна. Отмечается незначительная асимметрия треугольников талии, асимметрия таза влево. Ось позвоночника искривлена в левой фронтальной плоскости в поясничном отделе. Ось нижних конечностей правильная. Движения в суставах полные, безболезненные, отмечается гипермобильность надколенников. Поперечное плоскостопие, вальгусная деформация первых пальцев стоп.
Предварительный диагноз: «Остеопатии неясного происхождения. Сколиоз 1степени. Поперечное плоскостопие. Вальгусная деформация первых пальцев стоп».
Клинический анализ крови: гемоглобин 128г/л, лейкоциты 5,2×10 9 /л, палочкоядерные 1%, сегментоядерные 54%, эозинофилы 1%, лимфоциты 36%, моноциты 8%, тромбоциты 218×l0 9 /л, цветовой показатель 0,9%, скорость оседания эритроцитов 8мм/ч.
ЭКГ без особенностей.
Рентгенография голеней и коленных суставов 31.05.2017. Выявлена фиброзная дисплазия большеберцовых костей. По результатам КТ нижних конечностей от 01.06.2017 выявлена полиоссальная форма фиброзной дисплазии костей нижних конечностей.

Было принято решение провести дифференциальную диагностику между различными формами полиоссальной фиброзной дисплазии. На повторном приеме выяснились подробности анамнеза жизни, которые мать ранее скрывала. Выписки отсутствовали в амбулаторной карте и хранились у матери дома.
Продолжение анамнеза жизни и заболевания: в шестимесячном возрасте отмечалось изолированное телархе. В 2года двустороннее телархе, уплотнения молочных желез справа диаметром 3,5см, слева 3см. Половая формула Ма2Р0Ах0. За 12мес увеличение молочных желез повторялись 5раз, длительность прогресса- 3нед и регресса- 2нед. Консультирована и обследована эндокринологом в Институте детской эндокринологии ФГБУ «НМИЦ эндокринологии» Министерства здравоохранения России. В 4года у девочки появились менструальноподобные выделения. При УЗИ выявлена фолликулярная киста правого яичника. Проведена операция: удаления фолликулярной кисты (кистэктомия). После оперативного лечения признаки преждевременного полового созревания исчезли, темпы роста в норме, набухания молочных желез больше не отмечалось.
После анализа полного анамнеза жизни (включающего преждевременное половое развитие), а также данных рентгенографии и компьютерной томографии заподозрен синдром МОБ. Девочка направлена в Институт детской эндокринологии, где ранее лечилась.
Данные лабораторных и функциональных исследований.
Клинический анализ крови и мочи: норма.
Биохимический анализ крови: общий кальций- 2,27 (норма 2,02–2,6 ммоль/л), кальций ионизированный- 1,1 ммоль/л (норма 1,13–1,32ммоль/л), фосфор неорганический- 3,66ммоль/л (норма 0,86–1,56ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: норма.
Тиреоидный профиль: ТТГ 3,2мкМЕ/л (норма 0,32–5мкМЕ/л); Т4 общий 65нмоль/л (норма 39–155 нмоль/л); Т4 свободный 21нмоль/л (норма 13–30нмоль/л).
Гормональный профиль: пролактин 6,1нг/мл (норма 4–23нг/мл); соматомединС 423нг/мл (норма 95–473нг/мл); паратгормон 16нг/л (норма 8–24нг/мл); эстрадиол 24пг/мл (норма 22–30пг/мл); ФСГ 6,8мЕД/л (возрастная норма 1,5–4,0мЕД/л); ЛГ 1,75мЕД/мл (возрастная норма 0,5–4,6мЕД/л).
Сцинтиграфия с технецием: отмечается повышение накопления фармакологического препарата в области верхней трети правой бедренной кости (102%),в области средней трети правой большеберцовой кости (180%), правой таранной кости (106%).
Рентгенография черепа в боковой проекции- без патологии.
УЗИ щитовидной железы: общий объем 6,6 см 3 ; эхогенность средняя; структура паренхимы неоднородная, расширенные фолликулы. УЗИ других внутренних органов без особенностей.
Инструментальными и лабораторными методами исследования исключены заболевания паращитовидных желез, гипофиза, эпифиза и надпочечников. Подтвержден диагноз: «Синдром МОБ ».
Этиотропное лечение при полиоссальной фиброзной остеодисплазии пока не разработано.
Рекомендовано: щадящий ортопедический режим, прекращение занятий танцами, ношение ортопедических стелек, избегание физических нагрузок и травм, лечебная физкультура для укрепления мышц нижних конечностей, поясничной области, спины, плавание. Назначено медикаментозное лечение: альфакальцидол 0,25мкг 1раз в сутки, под контролем биохимического анализа антиостеопорозное средство (остеогенон 1таблетка 2раза в день в течение 1мес 2раза в год), регулятор фосфорно-кальциевого обмена (кальцемин Адванс 1таблетка 2раза в день в течение 1мес 4раза в год) .
Спустя 1год в результате нарушения режима девочка получила закрытый перелом средней фаланги 4-го пальца правой стопы в батутном центре. На рентгенограммах правой стопы в прямой и косой проекциях визуализируется патологический перелом средней фаланги 4-го пальца правой стопы, истончение кортикального слоя 2-го и 4-го пальцев, костные балки дифференцируются слабо. Произведена иммобилизация гипсовой повязкой на срок 4нед. По истечении данного срока на рентгенограммах в прямой и косой проекциях отмечается диастаз между отломками, замедленная консолидация, что характерно для синдрома МОБ.


Рис.3а, б.Рентгенограммы правой стопы: а- до иммобилизации, б- после 4нед иммобилизации
Планируется госпитализация для подбора дозы бисфосфонатов. В настоящее время оперативное лечение нецелесообразно, поскольку с наступлением пубертата возможно замедление или даже прекращение дальнейшего развития остеодисплазии. Рассмотреть возможность операции следует при кистозных образованиях, непосредственно угрожающих патологическим переломом.
1.При подозрении на синдром МОБ необходим тщательное изучение анамнеза, так как отсутствие полных данных в медицинской документации не является редкостью.
2.Критерии диагностики: кожные проявления- невусы цвета кофе с молоком, преждевременное половое развитие, фиброзная остеодисплазия. Поскольку значительно чаще встречаются неполные и стертые формы синдрома МОБ, заподозрить его следует при наличии только двух признаков.
3.Лабораторные исследования (гормоны сыворотки крови, биохимический анализ крови) могут быть без патологических изменений. Могут также отсутствовать аномалии надпочечников, гипофиза, признаки усиленного роста в длину, акромегалии ит.п.
4.Соотношение ФСГ:ЛГ>2,5 (в данном случае 3,9) говорит о вероятности повторного возниковения кисты яичника и необходимости наблюдения эндокринологом.
5.Необходимо периодческое рентгенографическое обследование для выявления прогрессирования фиброзных кист.
Список сокращений.
АКТГ- адренокортикотропный гормон.
Ax- оволосение подмышечных впадин.
КТ- компьютерная томография.
ЛГ- лютеинизирующий гормон.
ЛФК- лечебная физическая культура.
Ма- молочные железы.
МОБ синдром- синдром Мак-Кьюна- Олбрайта- Брайцева.
ОРВИ- острая респираторно-вирусная инфекция.
ПТГ- паратиреоидный гормон
Р- оволосение лобка.
Т4- тироксин.
ТТГ- тиреотропный гормон.
УЗИ- ультразвуковое исследование.
ФП- фармакологический препарат.
ФСГ- фоликуллостимулирующий гормон.
цАМФ- циклический аденозинмонофосфат.
Литература.
1. МаказанН.В. Роль нарушений пострецепторного сигналинга в развитии мультигормональной резистентности и автономной гиперфункции эндокринных желез у детей». – Дисс … канд. мед. наук.– М., 2017.
2. УвароваЕ.В. Обоснование использования фитопрепарата на основе Vitex agnus castus у девочек с преждевременным половым созреванием, ассоциированным с синдромом Мак-Кьюна-Олбрайта-Брайцева // Pediatric and Adolescent Reproductive Health.- 2017. - №6. - С.77–83.
3. ПлаксинаМ.И., Витебская А.В. Вариабельность симптомов при синдроме Мак-Кьюна- Олбрайта - Брайцева // Педиатрия.- 2016. - № 6 (123).
4. Синдром Олбрайта: причины, признаки, диагностика, как лечить // Синдром.info: Все о синдромах. - http://sindrom.info/olbrajta/
Синдром Олбрайта, или иначе псевдогипопаратиреоз - это очень редкое генетически детерминированное заболевание, которое проявляется нарушениями кальциевого и фосфорного обмена и, как следствие, патологией костной системы. Как правило, основные симптомы сопровождаются еще и отставанием в развитии - как умственном, так и физическом. Чаще заболевание встречается у женщин.
Этиология
Ученые считают, что синдром Олбрайта обусловлен интактностью клеток почек и костей к гормонам паращитовидных желез. Это связано с дефектом рецепторов. В почках нарушается процесс синтеза цАМФ (циклического аминазинмогофосфата), который является вспомогательным звеном в метаболических процессах, связанных с паратгормоном. Такие повреждения появляются при повреждении специфических генов.
Нарушение может быть на любом участке метаболической цепи. У одних больных поврежден сам рецептор, у других изменениям подвергаются белки клеточной мембраны, у третьих заболевание связано с недостаточной выработкой цАМФ. Все эти случаи связывает генетическая обусловленность патологии.
Патогенез
Синдром Олбрайта тесно связан с физиологией паращитовидных желез. В норме они регулируют уровень кальция и фосфора в крови, но при данном патологическом состоянии они равнодушны к колебаниям этих элементов, что может привести к возникновению вторичного гиперпаратиреоза. Но даже высокие уровни парагормона не стимулируют ускоренное элиминирование фосфора и цАМФ из крови (так как почки интактны к его воздействию) и в тоже время провоцируют изменения в структуре костей.
Как правило, наблюдается компенсаторное увеличение паращитовидных желез, пористость костей, появление в них кистозных образований, кальцификатов в коже, жировой ткани, мышцах, сердце, сосудах и конъюнктиве и роговице.
Клиника

Синдром Олбрайта проявляется практически так же, как и истинный гипопаратиреоз. У больных наблюдаются судороги, которые могу возникать как сами по себе, так и после воздействия раздражающих факторов (нервное напряжение, перепад температур, тяжелая физическая работа и т.д.). Кальцинаты, располагающиеся в коже и подкожно-жировой клетчатке, могут изъязвляться и доставлять дискомфорт пациентам. При этом больной будет обращаться за помощью не к эндокринологу или генетику, а к хирургу или дерматологу.
Такие люди, как правило, низкого роста, у них круглое одутловатое лицо, ожирение и укорочение пальцев. Наблюдаются ложные суставы там, где их быть не должно, отсутствует подвижность в тех местах, где она предполагается анатомически, нарушение конфигурации суставов и костей. Среди симптомов также рвота, кровь в моче, катаракта и изменения в зубной эмали.
Кроме того, из-за низкого уровня кальция в крови с детства у пациентов наблюдается снижение интеллекта и задержка развития.
Диагностика

Синдром Олбрайта - болезнь, которую довольно тяжело диагностировать у младенца, но в возрасте 5-10 лет появляются достаточно яркие проявления, которые не оставляют у врача сомнений в диагнозе. У детей наблюдаются множественные пороки развития костной системы, в крови снижен уровень кальция и повышен уровень фосфора и паратгормона.
Для подтверждения диагноза можно провести тест на количество фосфатов и цАМФ в моче. Для этого пациенту вводится 200 единиц паратгормона внутривенно и затем через 4 часа собирается порция мочи. Если достоверного повышения не наблюдается, то это может свидетельствовать о резистентности почечной ткани к паратгормону.
Этих признаков достаточно, чтобы поставить синдром Олбрайта. Диагностика может быть дополнена рентгенографией. Она дает возможность увидеть специфические изменения в костной системе и мягких тканях пациента.
Кроме паратгормона у людей с таким диагнозом регистрируют резистентность и к другим гормонам, поэтому женщинам проводят дифференциальную диагностику с синдромом Шерешевского-Тернера. Эти два заболевания имеют внешнюю схожесть, и для верификации диагноза врач должен назначить анализ на половой хроматин и дать направление на гинекологическое обследование и УЗИ органов малого таза.
Лечение и прогноз

После того как будет поставлен диагноз «синдром Олбрайта», лечение необходимо назначать, не откладывая, так как задержки усугубляют дефицит умственного развития. Детям назначают препараты кальция в достаточных суточных дозах, чтобы поддерживать нормальную концентрацию в сыворотке крови. Затем присоединяют к этому прием витамина Д в дозе, не превышающей 100 000 единиц в день.
Для корректировки лечения необходимо контролировать уровень кальция каждые 7 дней в течение первых двух недель, а затем раз в месяц до конца терапии. Когда оптимальная концентрация кальция в крови будет достигнута, можно проверять его раз в квартал.
Чтобы устранить симптомы вторичного гиперпаратиреоза, рекомендуется соблюдать диету со сниженным количеством фосфора. При обнаружении недостаточности других гормонов проводят заместительную и симптоматическую терапию.
Если лечение начато во время и терапия рациональная, то прогноз для жизни благоприятный. Но перед беременностью женщинам с таким диагнозом необходимо пройти консультацию у генетика.
Синдром Олбрайта – редкое генетическое заболевание с невыявленным происхождением. Передается по наследству и в основном характеризуется поражением костной системы. Чаще всего встречается у представительниц женского пола. Но бывает, когда заболеть могут и мальчики. Большинство пациентов с таким синдромом страдают физической и умственной отсталостью.
Название заболевание получило по фамилии ученого, который его открыл. Костная система начинает разрушаться под воздействием метаболизма кальция и фосфора. Паращитовидная железа выделяет паратгормон, у человека с синдромом Олбрайт резистентность тканей к паратгормону нарушена.
Симптомы синдрома Батлера Олбрайта
Выделяют три основных признака заболевания. При наличии даже двух из трех можно ставить такой диагноз.
- Очень раннее половое созревание в результате сильно завышенного гормонального фона. Менструация может начаться у девочек в первые месяцы после рождения. У мальчиков, страдающих синдромом Олбрайта, очень редко наблюдается раннее половое созревание. Изменение в гормональном фоне происходит из-за неправильной работы надпочечников, гипофиза и щитовидной железы. Этот симптом может проявляться в полной или неполной форме. При полной форме менструации начинаются очень рано, обычно они обильные и болезненные. В младенческом возрасте при этом синдроме проявляются и вторичные половые признаки, например, набухание молочных желез. При неполном проявлении менструации не бывает. Очень часто при синдроме Олбрайта у девочек возникают кисты на яичниках. У мальчиков при раннем половом созревание наблюдается увеличение члена и оволосение лобка.
- Фиброзная остеодисплазия. Это заболевание говорит о нарушении костной ткани, приводит к искривлению и деформации костей. Характеризуется асимметричностью. Самые распространенные признаки – хромота и искривление позвоночника. Чаще всего подвергаются изменению трубчатые кости. Рост скелета замедляется, это заметно даже у детей. У многих пострадавших от синдрома Мак-Кьюна наблюдается неравномерный рост костей черепа, что приводит к внешнему уродству. При изменении лица возможно проявление пучеглазия. После достижения возраста полового созревания костные изменения замедляются, и частота переломов сокращается.
- Изменения кожных покровов. Проявляется в повышенной пигментации. Кожа на туловище и бедрах покрывается желто-коричневыми пятнами с неровными краями. Пигментацию можно увидеть на пояснице, верхней части спины, ягодицах и на груди. Пятна появляются сразу же после рождения ребенка.

Лечение синдрома Олбрайта (Мак-Кьюна)
 Пациенты, страдающие синдромом Мак-Кьюна
, должны все время находиться под наблюдением узких специалистов (гинеколог, эндокринолог, травматолог, окулист).
Пациенты, страдающие синдромом Мак-Кьюна
, должны все время находиться под наблюдением узких специалистов (гинеколог, эндокринолог, травматолог, окулист).
Необходимо комплексное лечение. Так как болезнь характеризуется генетической мутацией, то необходимо в первую очередь избавляться от симптомов его проявления.
Показано обязательное гормональное лечение для поддержания эндокринной системы.
Необходимо не упустить момент, когда начнут деформироваться кости лица, чтобы предотвратить эти изменения во время болезни.
Иногда требуется и хирургическое вмешательство . Его назначают в том случае, когда из-за изменений в строении черепа возникает риск снижения слуха или зрения.
Во время болей используют специальные обезболивающие средства, содержащие небольшое количество бисфосфонатов.
Очень важно проводить профилактические меры по укреплению мускулатуры.
Для каждого пациента, страдающего болезнью Олбрайта, врач подбирает свое индивидуальное лечение, которое требуется соблюдать неукоснительно.
Необходимо постоянно контролировать количество кальция в организме. При начале проведения терапии такое обследование проводится раз в неделю, затем их частота сокращается до одного раза в месяц, пока не закончится основное лечение.
Очень важно во время лечения соблюдать диету, при которой количество продуктов, содержащих фосфор, урезается до минимума.
Следует заметить, что, женщина, страдающая этим синдромом , при решении забеременеть обязательно должна проконсультироваться с хорошим генетиком.
Болезнь Олбрайта (Мак-Кьюна) встречается у одного человека на 1 000 000. Специфического лечения для этого синдрома еще не изобрели, но не стоит терять надежды. Медицина не стоит на месте, а врачи помогут преодолеть неприятные моменты и будут держать пациента все время под наблюдением.
Catad_tema Педиатрия - статьи
Псевдогипопаратиреоз (наследственная остеодистрофия Олбрайта): сложности дифференциально-диагностического поиска. Клиническое наблюдение
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., И.В. Шулякова, невролог, к. м. н.,
обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Минздрава РФ, г. Москва
Ключевые слова:
дети, псевдогипопаратиреоз, наследственная остеодистрофия Олбрайта, ожирение, гипокальциемия, диагностика, резистентность к паратиреоидному гормону.
Keywords:
children, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Псевдогипопаратиреоз (греч. pseudes – ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, синдром «яванской курицы») – редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Заболевание описано впервые американским врачом-эндокринологом Albright F. в 1942 году . Распространенность заболевания составляет 7,9 на 1 млн человек .
ГЕНЕТИЧЕСКИЕ ДАННЫЕ
Псевдогипопаратиреоз (ПГП) – генетически гетерогенное заболевание. Данные о типе наследственной передачи противоречивы: как X-сцепленный доминантный , так и аутосомно-доминантный, аутосомно-рецессивный типы . В большинстве случаев развитие наследственной остеодистрофии Олбрайта связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1 (Patten et al., 1990), кодирующего белок Gs-альфа, связанного с рецептором паратиреоидного гормона (ПТГ) . Подобный фенотип выявлен и у больных с интерстициальной делецией длинного плеча хромосомы 2 локуса 2q37 .
ПАТОГЕНЕЗ
В основе патогенеза псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию парат-гормона в результате дефекта комплекса «специфический циторецептор – паратгормон – аденилатциклаза», что нарушает процесс образования в почках циклического 3"-, 5"-аденозинмонофосфата (цАМФ), являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип 1А псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип 1B псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз 2-го типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней к паратгормону .
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
В настоящее время выделяют 4 клинические формы патологии: типы 1А, 1В, 1С и 2. Знание их клинико-биохимических особенностей и данных генетических исследований позволяет провести дифференциальную диагностику в рамках самой нозологической формы.
Общими признаками, позволяющими заподозрить заболевание, являются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей (фото 1), брахидактилия (фото 2), круглое «лунообразное» лицо (фото 3). Иногда наблюдаются экзостозы и аплазия зубов.
Фото 1.
Внешний вид ребенка с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост за счет укорочения нижних конечностей)
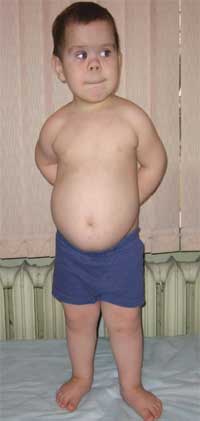
Фото 2.
Особенности костной системы у больного
с остеодистрофией Олбрайта
(брахидактилия – укорочение пальцев)
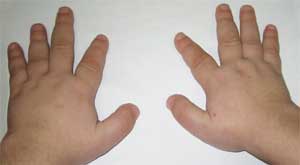
Фото 3.
Особенности фенотипа ребенка
с остеодистрофией Олбрайта
(круглое «лунообразное» лицо)

Патогномоничным признаком считается резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), вследствие чего II пальцы на кистях и стопах оказываются длиннее остальных, а при сжатии кисти в кулак отсутствуют выпуклости в области IV и V пястно-фаланговых суставов – так называемый брахиметафалангизм. Выявляются также короткие широкие фаланги, утолщение свода черепа и деминерализация костей (остеопороз), ожирение .
Умственная отсталость (чаще умеренной степени выраженности) обнаруживается примерно у 20% больных. По данным некоторых авторов , олигофрения встречается в 70% случаев при наличии гипокальциемии и в 30% случае при нормокальциемии. Психические процессы у больных замедлены. В неврологическом статусе нередко отмечаются моторная неловкость, невротические реакции: страхи, тревога, беспокойство, плохой сон, повышение рефлексов, судороги, носящие тетанический характер и обусловленные гипокальциемией, иногда судорожные пароксизмы. Описаны также миопатические симптомы: мышечная утомляемость, мышечная слабость. Часто наблюдаются экстрапирамидные нарушения: хореиформные гиперкинезы, атетоз, лицевой гемиспазм, паркинсонизм, в отдельных случаях имеют место эпилептические пароксизмы, мозжечковые симптомы: атаксия, нарушение координации.
Нередко определяются кальцификация мягких тканей, подкожные кальцификаты (грудь, живот, пяточные сухожилия), при гистологическом исследовании которых – osteoma cutis (Izraeli et al., 1992), мозге (базальные ганглии). Важно отметить, что кальцификаты могут быть уже при рождении. Вследствие гипокальциемии обычно развивается катаракта и возникают дефекты эмали зубов.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1А
имеет аутосомно-доминантный тип наследования. Ген псевдогипопаратиреоза типа 1А – GNAS1 – локализован на длинном плече хромосомы 20, в локусе 20q13.2. Развитие заболевания связано с дефицитом гуанин-нуклеотид-связывающего белка (Gs-белок). При этом ПТГ, связываясь с рецепторами тканей-мишеней, не способен активизировать циклический аденозинмонофосфат (цАМФ) и вызвать тканевой ответ. Вероятно, подобный механизм лежит в основе развития нечувствительности тканей других органов и эндокринных желез (гипофункция щитовидной железы, гонад, гипофиза, сахарный диабет, а также сниженный ответ печени на введение глюкагона), наблюдаемой при псевдогипопаратиреозе типа 1А. При данном типе патологии не наблюдается характерной для нормы повышенной экскреции цАМФ с мочой в ответ на экзогенное введение ПТГ. Заболевание диагностируется чаще в возрасте 5–10 лет. У больных наблюдаются низкий рост, короткая шея, круглое лицо, укорочение метакарпальных и метатарзальных костей (чаще укорочение IV и реже II пальцев) – так называемый брахи-метафалангизм. Отмечаются кальцификация мягких тканей, подкожные кальцификаты, которые могут выявляться уже при рождении; нередко наблюдается одновременное вовлечение других эндокринных желез: щитовидной железы (гипофункция), гонад, поджелудочной железы (сахарный диабет). Вследствие гипокальциемии нередко развиваются катаракта и дефект эмали зубов. В качестве дифференциально-диагностического теста отличия ПГП типа 1А от гипопаратиреоза: отсутствие клинического эффекта от парентерального введения ПТГ в виде подъема уровня кальция в крови и увеличения почечной экскреции фосфора с мочой (фосфатурический эффект).
При биохимическом исследовании выявляются гипокальциемия, гиперфосфатемия, увеличение уровня паратиреоидного гормона в крови, гипофосфатурия. Уровень Gs-белка в крови снижен. При рентгенологическом исследовании костной системы обнаруживаются укорочение метакарпальных и метатарзальных костей, генерализованная деминерализация, утолщение костей свода черепа.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1B
имеет аутосомно-доминантный тип наследования, однако не исключен доминантный, сцепленный с Х-хромосомой тип наследования. Необходимо иметь в виду наблюдающуюся иногда неполную пенетрантность гена болезни и возможность скрытого носительства патологии. Поэтому рекомендуется клиническое (выявление субклинического течения болезни) и биохимическое обследование (определение уровней кальция, фосфора, ПТГ крови) предполагаемых носителей заболевания. ПГП типа 1В обусловлен дефицитом тканевых рецепторов к паратиреоидному гормону в органах-мишенях и ограниченной резистентностью к паратгормону. Клиническая картина сходна с клиникой типа 1А, но отсутствует поражение других эндокринных желез, реже встречается остеодистрофия.
У больных отсутствует реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой, однако, в отличие от типа 1А, уровень Gs-белка в крови нормален. Женщины поражаются чаще мужчин, однако тяжесть заболевания может быть одинаковой как у мужчин, так и у женщин.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1С
некоторые авторы отождествляют с псевдо-псевдогипопаратиреозом (ППГП), описанным Albright F. в 1952 году. Характеризуется свойственной ПГП клинической картиной, однако уровни кальция, фосфора в крови и моче остаются в пределах нормы. Показатели ПТГ и Gs-белка в крови также сохраняются на нормальном уровне. У некоторых больных с ПГП типа 1С обнаруживаются делеции de novo на хромосоме 2 . Не исключено, что этот вариант болезни является подтипом ПГП типа 1А.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 2
клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Не исключено существование и аутосомно-доминантных форм патологии. Патогенез развития связан с внутриклеточной резистентностью к цАМФ. ПТГ при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на ПТГ в виде увеличения экскреции цАМФ. Внутриклеточная нечувствительность к цАМФ, однако, не позволяет осуществиться полной реализации действия ПТГ. При этом сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой. Высказывается мнение, что ПГП типа 2 может быть связан с дефицитом витамина D .
Таким образом, выделенные типы ПГП клинически характеризуются пониженной чувствительностью органов-мишеней к ПТГ, однако различаются патогенетическими механизмами формирования нечувствительности тканей.
ДИАГНОСТИКА
Лабораторным дифференциально-диагностическим тестом может служить характер почечной экскреции цАМФ в ответ на введение ПТГ: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие – при типе 1. Диагноз подтверждается обнаружением сниженного уровня гуанин-нуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень ПТГ повышен; при 1C-типе уровень ПТГ в норме, что дало основание для названия «псевдогипопаратиреоз». При рентгенологическом исследовании костной системы обнаруживаются укорочение пястных и плюсневых костей, нередко генерализованная деминерализация (остеопороз), утолщение костей свода черепа. В дерматоглифическом рисунке отмечается смещение осевого ладонного трирадиуса.
Критерии диагноза:
- низкий рост;
- круглое лицо;
- задержка нервно-психического развития;
- скелетные аномалии;
- низкое содержание кальция в сыворотке крови;
- высокий уровень паратиреоидного гормона в крови;
- снижение экскреции с мочой фосфатов и цАМФ.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. В настоящее время применяют активные метаболиты витамина D – оксидевит, 1-альфа-Д3, кальцитрин и др. в дозе 1–2 мкг/сутки с положительным результатом (увеличение содержание кальция в крови, уменьшение проявлений судорожного синдрома). Эффективен также тахистин (0,5–1,5 мг/сутки). Данный препарат увеличивает всасывание кальция в кишечнике и тем самым способствует повышению уровня кальция в крови. Противосудорожная терапия используется как дополнительное лечение. На интеллектуальное развитие лечение не оказывает заметного действия, но наряду с уменьшением симптомов судорожного синдрома наблюдается регресс неврологических проявлений (подкорковых нарушений, хореиформных гиперкинезов, атетоза и др.). Во избежание передозировки препаратов витамина D необходим контроль концентрации кальция в крови каждые 3–7 дней в течение первых 2 недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца. Диета с ограничением фосфора помогает нормализовать как концентрацию фосфора, так и содержание кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами.
Лечение паратгормоном неэффективно. Для купирования судорожных приступов внутривенно вводят 10%-ный раствор кальция хлорида или кальция глюконата; внутрь – 5–10%-ный раствор кальция хлорида по 1 столовой ложке 3–4 раза в день: кальция глюконат, кальция лактат – до 10 г в день.
ПРОГНОЗ для жизни определяется выраженностью судорожного синдрома.
ПРОФИЛАКТИКА болезни основывается на данных медико-генетического консультирования.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в гене GNAS1 на хромосоме 20. Разрабатываются способы пренатальной диагностики заболевания в целом и отдельных его типов.
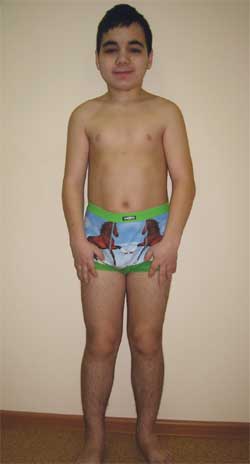
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ Мальчик Г., 14,5 лет (фото 4), поступил в Научно-исследовательский клинический институт педиатрии с диагнозом: дегенеративное заболевание нервной системы? врожденная наружная гидроцефалия; симптоматическая эпилепсия; наследственный синдром? болезнь накопления? метаболическая энцефалопатия; субклинический гипотиреоз; низкорослость смешанного генеза; когнитивные нарушения.
Жалобы при поступлении на интенсивные приступообразные головные боли, локализующиеся в лобной области и сопровождающиеся рвотой, которая приносит облегчение, снижение памяти и успеваемости в школе, судорожные приступы, во время которых происходит подергивания в правой руке.
Фото 4.
Ребенок Г., 14,5 лет, с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост, укорочение конечностей, брахидактилия)
Анамнез семейный: родители – армяне по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии – не отмечалось. Сибс, сестра 17 лет – со слов – здорова.
Анамнез жизни и заболевания:
мальчик от 2-й беременности, протекавшей без особенностей, роды вторые, в срок, физиологические, масса при рождении – 3100 г, длина – 51 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. Ухудшение состояния на 3-и сутки – судороги неонатальные, купированы в роддоме. Ранний постнатальный период – без особенностей. Отмечалась незначительная темповая задержка моторного развития на первом году жизни, самостоятельная ходьба с 1 года 3 мес. В связи с чем наблюдался неврологом с диагнозом: органическое поражение ЦНС; врожденная гидроцефалия; неонатальные судороги; фебрильные судороги в анамнезе.
Получал диакарб, финлепсин. Дебют приступов с 1 года 11 мес. – асимметричные, тонические в виде напряжения правой руки и ноги, с заведением глаз, до 2 мин., без потери сознания, частые до 10 эпизодов в сутки. Получал депакин нерегулярно. На фоне самостоятельной отмены – однократный тонический статус. В 2 года проведена по месту жительства КТ головного мозга, где выявлены единичные очаги демиелинизации в затылочных долях.
Консультирован нейрохирургом, рекомендовано консервативное лечение. С 3 лет отмечается задержка психоречевого развития, рекомендовано наблюдение психиатра.
С 4–5 лет родители стали отмечать деформацию и укорочение пальцев стоп и кистей, в особенности II–IV пальцев симметрично на руках и ногах, снижение ростовых показателей. В 8 лет заключение логопеда – общее нарушение речи 2–3-го уровня, рекомендовано обучение в специализированной школе. В этом же возрасте осмотр генетиком по месту жительства, заключение: наследственная болезнь обмена? рекомендовано исследование аминокислот крови, изменений не было выявлено; окончательное заключение: данных за наследственное заболевание обмена не выявлено; гипохондроплазия; рекомендовано лечение невролога и эндокринолога.
Таблица.
Профиль психического развития ребенка Г., 14,5 лет (IQ = 68)

В возрасте 8 лет консультирован эндокринологом по поводу задержки роста и развития. При рентгенологическом исследовании кистей рук отмечены особенности: средние, основные фаланги и пястные кости укорочены, утолщены; диагноз рентгенолога – ахондроплазия.
Неоднократно обследуется по месту жительства в неврологическом стационаре. В 12 лет появились судорожные приступы без потери сознания с подергиванием правой руки, носящие серийный характер, назначена противосудорожная терапия (депакин), частота приступов значительно сократилась. В 13 лет проведена МРТ головного мозга с контрастированием – симметричные изменения в основании височных долей на уровне ядер в виде повышения МР-сигнала, что характерно для токсических (марганец) или метаболических (медь, железо) энцефалопатий.
Вновь осмотрен в возрасте 13 лет 3 мес. эндокринологом, при исследовании тиреоидного профиля выявлено повышение тиреотропного гормона (ТТГ), диагностирован субклинический гипотиреоз, назначен L-тироксин.
При анализе амбулаторной карты ребенка и документации по месту жительства исследование кальция и фосфора проводилось однократно, в возрасте 1,5 лет, отмечалась гипокальциемия, но по данному поводу дообследование не проводилось. Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Москву, в Научно-исследовательский клинический институт педиатрии, с целью уточнения диагноза.
Данные объективного исследования:
Рост – 143 см, масса – 43 кг.
Физическое развитие очень низкое, гармоничное, телосложение диспропорциональное за счет укорочения конечностей. Sds роста соответствует –2,8 отклонениям от нормы (норма –2+2).
Особенности фенотипа: круглое лицо, короткая шея, антимонголоидный разрез глазных щелей, широкое переносье, высокий лоб, брахидактилия, укорочение IV и V пястных и плюсневых костей (фото 5). По внутренним органам – без особенностей. Половое развитие – Tanner III–IV стадия (что соответствует возрасту).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови: общий кальций – 1,39 (норма 2,02–2,6 ммоль/л), кальций ионизированный – 0,61 (норма 1,13–1,32 ммоль/л), фосфор неорганический – 3,66 (норма 0,86–1,56 ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: почечная экскреция фосфатов снижена – 11,5 ммоль/л (норма 19–32 ммоль/л).
Тиреоидный профиль: ТТГ – 11,75 (норма 0,4–4,0 мкМЕ/мл), свободный Т4 – 0,49 (норма 1,0–1,8 нг/дл).
Паратиреоидный гормон – 499 (норма 12– 65 пг/мл), СТГ – 7 нг/мл (норма 7–10 нг/мл), соматомедин-С – 250 нг/мл (норма 88–360 нг/мл).
УЗИ внутренних органов – без особенностей.
ЭКГ – миграция суправентрикулярного водителя ритма на фоне регулярной ЧСС 71– 80 уд/мин. Неполная блокада правой ножки пучка Гиса. Нарушение процесса реполяризации в миокарде задней стенки левого желудочка (снижение з.Т III, аVF).
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника 1-й степени, выраженный остеопороз.
R-графия кистей рук с захватом предплечий – укорочение и расширение концевых и средних фаланг. Костный возраст – 13,5– 14 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – МР-картина множественных субкортикальных очагов повышенного МР-сигнала в лобных долях, наружная компенсированная гидроцефалия с атрофией вещества головного мозга.
МСКТ головного мозга – симметричные участки обызвествления лентиформных ядер. Диффузные гиперденсивные участки в таламусах, хвостатых ядрах с участком обызвествления справа. Множественные точечные обызвествления покровных мягких тканей черепа.
Аудиограмма – без патологии.
ДНК-диагностика в гене GNAS1 – в работе.
Консультации специалистов:
Эндокринолог – наследственная остеодис-трофия Олбрайта типа 1А (псевдогипопара-тиреоз). Первичный гипотиреоз, неполная медикаментозная компенсация.
Окулист – катаракта полная вторичная. Рекомендовано оперативное лечение.
Психолог – когнитивные нарушения (психологический профиль ребенка представлен в табл.).
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований (гипокальциемия, гиперфосфатемия, гипофосфатурия, повышение паратиреоидного гормона крови), кальцинаты в веществе головного мозга, наличие катаракты, гипотиреоза), поставлен диагноз: наследственная остеодистрофия Олбрайта 1А-типа (псевдогипопаратиреоз). Рекомендовано проведение ДНК-диагностики - поиск мутаций в гене GNAS1.
Лечение: ребенку рекомендован прием эутирокса в дозе 100 мкг/сутки; активный метаболит витамина Д - альфа-Д3 («Тева») в дозе 2 мкг/сутки; кальций («Сандоз») 2000 мг/сутки; постоянный прием противосудорожной терапии - финлепсин 800 мг/сутки под наблюдением невролога-эпилептолога; занятия с логопедом-дефектологом и психологом; энерготропная терапия (Элькар и коэнзим Q10 в возрастных дозах). Контроль показателей фосфорно-кальциевого обмена, уровня паратгормона.
Таким образом,
представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, важность своевременного исследования простых биохимических параметров (при эпилепсии обязателен неоднократный скрининг показателей фосфорно-кальциевого обмена), исходы поздней диагностики генетически детерминированного заболевания, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждение повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии. Список литературы находится в редакции.



