Albright syndrom. Albrights syndrom: årsager, symptomer, behandling. Denne sygdom forekommer hovedsageligt hos piger
McCune-Albright syndrom er en heterogen sygdom, som uafhængigt blev beskrevet af den amerikanske børnelæge D. J. McCune og endokrinolog F. Albright i 1936-1937. Sygdommen er sjælden, medfødt, men ikke arvelig, ledsaget af stofskifteforstyrrelser, tegn på for tidlig seksuel udvikling og skeletdeformiteter. De første kliniske manifestationer er bemærket i den tidlige barndom eller ungdom i form af udviklingsforstyrrelser, polyostotisk fibrøs knogledysplasi, café au lait hudpigmentering og endokrine abnormiteter, herunder, Cushings syndrom , .
Albright McCunes syndrom diagnosticeres cirka 10 gange oftere hos piger end hos drenge. Wikipedia bemærker, at hyppigheden af forekomst i den menneskelige befolkning er 1 pr. 100 tusind eller pr. million individer.
Patogenese
Som et resultat af en postzygotisk genmutation GNAS1 involveret i cellulær proteinsignalering GS-a, der sikrer koblingen af luteiniserende og follikelstimulerende hormonreceptorer med molekyler adenylatcyclase i de cytologiske strukturer i æggestokkene. Den konstante indflydelse af mutantproteinet aktiverer adenylatcyclase og øger intracellulært lejr , og stimulerer også øget sekretion af steroide kvindelige kønshormoner af follikulære østrogen-producerende cyster, hvilket forårsager for tidlig seksuel udvikling selv i fravær af. Det samme billede observeres og "autonom produktion" observeres med skjoldbruskkirtelhormoner, og. Progressionen af patologi er forårsaget af dannelsen af celler, der allerede indeholder mutante proteiner.
Derudover fører en stigning i niveauet af cAMP til en stigning i produktionen og dannelsen af lentigeniske pletter, fokal hyperpigmentering på baggrund af normale værdier og melanotropin .
Fibrocystisk dysplasi som et resultat af øget forkalkning under påvirkning af østrogener, udføres det ved at erstatte normale knoglestrukturer og dannede forkalkninger med fibrøse masser, mens balancen mellem processerne med knogleresorption og regenerering, spredning af mesenchymale stromaceller forstyrres. Dannelsen af foci af ødelæggelse og pseudotumorer observeres.
Klassifikation
Albrights sygdom kan være anderledes, afhængigt af sværhedsgraden af sygdommen skelnes de:
- skjult sygdom - uden tegn på knogle- eller endokrin patologi samt uden symptomer på usædvanlig pigmentering;
- ufuldstændige former for syndromet for eksempel i processen med patogenese, der involverer knoglevæv, er endokrine ændringer mulige og pseudohypoparathyroidisme , men ingen hudmanifestationer;
- pseudohypoparathyroidisme eller arvelig Albrights osteodystrofi - en sygdom, der også påvirker knoglevæv, men mekanismen og ætiologien er forskellig - patologien er forårsaget af nedsat resistens over for parathyreoideahormon , metabolisme af calcium og phosphor; pseudohypoparathyroidisme manifesterer sig også i barndommen og kommer til udtryk i form af kort statur, rundt ansigt, mental retardering, hypogonadisme , etc.

Årsager
Albright McCunes syndrom opstår som følge af genetiske lidelser i de tidlige stadier af embryogenese. Arvstypen er endnu ikke fastlagt alle beskrevne tilfælde var sporadiske.
Symptomer
Det kliniske billede af sygdommen manifesterer sig i form af et helt sympatokompleks, der består af:
- for tidlig gonadotropin-uafhængig seksuel udvikling, der udvikler sig senere og langsommere end i andre former for tidlig pubertet;
- pletvis hudpigmentering - lentigo , pletter med klare grænser af lysebrun farve er normalt til stede fra fødslen på brystet, ryggen, lænden, hofterne og på steder med knogledeformiteter;
- endokrine lidelser, der forårsager uforklarligt vægttab, øjensymptomer hyperthyroidisme ;
- polyostotisk fibrøs osteodysplasi , og, der ofte involverer lange knogler i processen, manifesterer lidelser sig i form af skæmmede ansigtstræk, en mere forlænget kropsbygning, buede og asymmetriske, forkortede håndled, som gør det vanskeligt at bevæge sig under gang, forårsager halthed, stærke smerter og øger sandsynlighed for patologiske frakturer.

De første tegn på for tidlig pubertet begynder med livmoderblødning, som ikke er cyklisk og skyldes kortvarige stigninger i koncentrationerne af østrogen og gonadotrope hormoner i blodbanen. Sådanne udsving fremkalder dannelsen af cyster i æggestokkene. Børn i alderen 5-10 år oplever tidlig brystvækst og gynækomasti . Dens blide ætiologi fører ikke til dannelsen af sekundære seksuelle karakteristika - McCune-Albright-Braitsev syndrom forårsager ikke tidlig seksuel hårvækst eller figurdifferentiering. Ægte pubertet er mulig efter pubertetens afslutning.
Test og diagnostik
Hvis der er mistanke om McCune Albrights syndrom, udføres følgende:
- Røntgenundersøgelser, der afslører pseudocyster , segmental skade på nogen dele af skelettet - oftest de nedre ekstremiteter, i mere sjældne tilfælde - den øvre skulderbælte og kraniet;
- Ultralyd af bughulen kan opdage;
- laboratorietests for at påvise øgede niveauer af alkalisk fosfatase i blodbanen, nedsatte niveauer af gonadotropiner i urinprøver og øget udskillelse luteiniserende hormon , 17-hydroxy Og 17-hydroxy kortikosteroider .
Behandling
Behandling er ordineret symptomatisk, det har sine egne vanskeligheder afhængigt af de individuelle egenskaber og det kliniske billede. Analoger GnRH har ikke en terapeutisk effekt, da processen med østrogenudskillelse ikke fremkaldes af et overskud af gonadotropiner. For at stoppe processen med gonadal steroidogenese hos mænd, og kan være effektiv. Under behandlingen medroxyprogesteronacetat mulig udvikling hypocalcæmi Derfor, hvis processen med udvikling af tidlig pubertet er ledsaget af alvorlige multiple læsioner af knoglevæv, skal terapi ordineres med forsigtighed.
For undertrykkelse hyperøstrogenisering og skelet forlængelse, medicin kan ordineres. For at modvirke knogledysplasi bør følgende administreres:
- bisfosfonater ;
- osteoklastantagonisthæmmere.
Patienter skal regelmæssigt, hver 3. måned. besøge en endokrinolog for at rette problemer med skjoldbruskkirtlen og andre kirtler.
Lægerne
Lægemidler
- – på trods af, at det er bedre kendt som et svampedræbende lægemiddel, er det i store doser effektivt til Kushina-lignende manifestationer. Den daglige dosis, opdelt i 2-3 doser, kan nå op på 1200 mg.
- Medroxyprogesteron – har androgene, antitumor- og anabolske virkninger. Forskellige indgivelsesveje i kroppen er mulige i doser individuelt bestemt af den behandlende læge.
- Pamidronsyre – lægemidlet er i stand til at forhindre osteolytisk knogleødelæggelse. Kan indgives intravenøst som bolus, men bør ikke kombineres med behandling med andre bisphosonater.
- – et bisfosfonat, der fremmer dannelsen af knogler med normal histologisk struktur, deres genopretning og forøgelse af mineraltæthed. Lægemidlet skal tages oralt - 1 tablet om ugen.
Procedurer og operationer
- Ovariektomi – fjernelse af æggestokkene kan anbefales i tilfælde af overdreven progression af hyperpubertal udvikling.
- Kirurgisk rekonstruktion af skelet- og kranieknogledeformiteter.
Vejrudsigt
I de senere år er folk begyndt at tale om Albrights McCune-syndrom og problemerne med dets behandling mere og oftere, og folk, der lider af denne sygdom, er begyndt at give sig selv til kende og afsløre de særlige forhold ved hverdagen og det sociale liv i personlige blogs og videoer . Takket være moderne teknologier, på trods af patologiens uhelbredelighed, er livskvaliteten for sådanne mennesker steget betydeligt, nu kan de finde en måde at udtrykke sig på og endda optræde i film, for eksempel som den uruguayanske skuespiller Mauricio Saravia.
Liste over kilder
- McCune. D. J. Osteitis fibrosa cystica: tilfældet med en ni år gammel pige, der også udviser tidlig pubertet, multipel pigmentering af huden og hyperthyroidisme. American Journal of Diseases of Children, Chicago, 1936; 52: 743-747.
- Mandel B. R. Fundamentals of moderne genetik. Tutorial. M: Flinta, 2017. – 305 s.
- Kulakov V.I., Manukhina I.B., Savelyeva G.M. Gynækologi: national. hænder M.: GEOTAR-Media, 2011.- 461 s.
Disse oplysninger er beregnet til sundheds- og farmaceutiske fagfolk. Patienter bør ikke bruge denne information som medicinsk rådgivning eller anbefalinger.
Et tilfælde af McCune-Albright-Braitsev syndrom
Traumatolog-ortopæd af den højeste kategori Bagrinovskaya Irina Leibovna
Moskva Børnebyklinik nr. 110
Nøgleord: hyperpigmentering / piger / børn / lentigo / mutation / patologisk fraktur / polyostotisk osteodysplasi / for tidlig seksuel udvikling / McCune-Albright-Braitsev syndrom / follikulær cyste.
McCune-Albright-Braitsev syndrom (McCune-Albright-Braitsev syndrom) eller Albright-McCune-Sternberg syndrom, som angivet under kode Q78.1 i den 10. internationale klassifikation af sygdomme. Den første indikation af denne sygdom tilhører den russiske og sovjetiske kirurg V.R. Braitsev, der i 1907 beskrevet fibrocystisk sygdom i kæben ( dysplasi fibrosa polycystia) hos børn med alderspletter. Syndromet fik dog først sit navn efter udgivelserne af den amerikanske børnelæge D.J. McCune i 1936. og den amerikanske endokrinolog F. Albright i samarbejde med patolog W. Sternberg i 1937.
Kliniske manifestationer af syndromet:
– asymmetriske hyperpigmenterede café-au-lait-pletter (lentigo), sædvanligvis på huden på brystet, ryggen, lænden og lårene;
- endokrinopatier, oftest for tidlig seksuel udvikling;
– polyostotisk fibrocystisk osteodysplasi.
MRD-syndrom er forårsaget af en somatisk mutation af GNASI-genet, placeret på den lange arm af kromosom 20, i de tidlige stadier af embryogenese, og er derfor ikke nedarvet. Som et resultat dannes kloner af mutante celler. GNASI-genet koder især for α-underenheden af G-protein (Gαs), som tjener som mellemled i omdannelsen af cyklisk adenosinmonofosfat (cAMP), som regulerer det menneskelige hormonsystem.
Med en stigning i niveauet af cAMP stiger produktionen af melanin med normale værdier af ACTH og melanostimulerende hormon, som et resultat af hvilke lentiginøse pletter dannes.
Da Gαs tjener som en mediator i signaltransmission fra LH og FSH, udviser mutante celler i gonaderne kraftigt øget autonom reaktivitet, hvilket fører til forekomsten af follikulære østrogen-producerende cyster og for tidlig seksuel udvikling hos piger. Mutante celler er ujævnt fordelt i kønskirtlerne. Hos piger kan de lokaliseres i en eller to æggestokke, hvilket forårsager den periodiske dannelse af ensidige eller bilaterale aktive cyster med deres efterfølgende regression.
Active Gαs initierer en ubalance mellem knogleresorption og regenerering, proliferation af umodne stromale celler, hvilket fører til udskiftning af normal knoglestruktur med fibrøse masser og dannelse af ødelæggelsesfoci i form af cyster.
Ved at mediere virkningerne af parathyreoideahormon (PTH) er Gαs involveret i at opretholde knoglehomeostase og calcium-phosphor-metabolisme. PTH er hovedfaktoren ansvarlig for homeostasen af calciummetabolisme. Calcium vaskes ud af knogler og aflejres i form af forkalkninger i subkutant fedt, muskler, indre organer og endotelet i arterielle kar. Over tid erstattes knoglevæv af fibrøst væv. Skeletabnormiteter hæmmer patienternes mobilitet og forårsager alvorlig smerte. Et fald i D-vitamin i blodet fører til forstyrrelse af optagelsen af calcium i tarmen. Hypokalcæmi stimulerer endnu større frigivelse af PTH til blodet og øget udvaskning af calcium fra knoglerne, hvilket fører til udvikling af svær osteomalaci.
Efterhånden som kroppen vokser og vægtbelastningen øges, bliver det patologiske område af knoglen bøjet. Hos en række patienter bliver lårbenet deformeret og får som følge af patologiske frakturer form som en hyrdeskurk. Hos nogle patienter fører alvorlig knogledeformation til invaliditet.
Ved tilstedeværelse af mutante celler i andre kirtler i det endokrine system kan multinodulær struma, hypofosfatemisk rakitis, Cushings syndrom, akromegali osv. forekomme. Når kardiomyocytter er beskadiget, observeres takykardi. Mulige læsioner i mave-tarmkanalen, såsom kolestatisk hepatitis, gastrointestinale polypper, gastrointestinal refluks.
Begge køn er modtagelige for denne sygdom, men den forekommer 2 gange oftere hos piger end hos drenge. For tidlig pubertet ses 9 gange oftere hos piger end hos drenge.
MRD-syndrom er en sjælden (forældreløs) sygdom. Hyppigheden af forekomst i verden varierer fra 1:100000 til 1:1000000 i den almindelige befolkning.
Forløb og prognose. Sygdommen er klinisk heterogen. Sammen med tilfælde af den klassiske triade af symptomer er der atypiske og ufuldstændige former for syndromet. Sværhedsgraden af symptomer og sværhedsgraden af sygdommen er også meget varierende. Knoglevævspatologi skrider frem med alderen, men med slettede former bremses progressionen med begyndelsen af puberteten. Udviklingen af knoglepatologi alene er 30-40 gange hyppigere end det fulde symptomkompleks.
Klinisk observation. Pigen V., 12 år gammel, blev indlagt hos en ortopædisk traumatolog på Moscow Children's City Clinic nr. 110 den 31. maj 2017.
Ved indlæggelse, klager over smerter i benene, forværring efter lange gåture eller balsaldans. Ifølge patienten er smerten så stærk, at det er umuligt at holde ud, at hun er nødt til at stoppe op og sætte sig ned nogen steder. Betragter sig selv som syg i 3 måneder.
Familiehistorie: forældre er russiske efter nationalitet, ikke beslægtet med blod. Der var ingen tilfælde af psykiske eller neurologiske sygdomme eller udviklingsforsinkelser i stamtavlen. Sibs, en 15-årig søster, er rask ifølge sin mor.
Livshistorie: barn fra anden graviditet, fuldbåren fødsel, fysiologisk, fødselsvægt 3500 g, højde 51 cm, skreg med det samme, Apgar score 8/9 point. Begyndte at holde hovedet på 2 måneder, siddende på 7 måneder, gå selvstændigt på 1 år, amning indtil 5 måneder. Vaccineret efter alder. Infektionssygdomme: ARVI, skoldkopper.
Objektive forskningsdata. Højde 158 cm, vægt 40 kg. Blodtryk 120/75 mmHg, puls 78/min. Generel stand er tilfredsstillende. Funktionel status: gåture, gang er ikke svækket. Fysikken er korrekt, normostenisk. På højre og venstre skuldre, på kroppens laterale overflade til højre, er der enkelte små lysebrune lentiginøse pletter med ujævne kanter med en diameter på op til 20 mm. Subkutant fedtvæv er moderat udviklet og jævnt fordelt. Synlige slimhinder er rene, lyserøde og fugtige. Perifere lymfeknuder er enkelte, små, mobile, smertefrie. Muskelmassen er tilstrækkeligt udviklet. Der er en let asymmetri i taljetrekanterne og asymmetri i bækkenet til venstre. Spinalaksen er buet i venstre frontalplan i lænderegionen. Aksen for underekstremiteterne er korrekt. Bevægelser i leddene er fuldstændige, smertefrie, hypermobilitet af knæskallerne er noteret. Tværgående fladfod, valgus deformitet af de første tæer.
Foreløbig diagnose: ”Osteopatier af ukendt oprindelse. Skoliose 1. grad. Tværgående fladfod. Valgus deformitet af de første tæer."
Klinisk blodprøve: hæmoglobin 128 g/l, leukocytter 5,2×10 9 /l, bånd 1 %, segmenteret 54 %, eosinofiler 1 %, lymfocytter 36 %, monocytter 8 %, blodplader 218×10 9 /l, farveindeks 0 . 9 %, 8 mm/t.
EKG uden nogen funktioner.
Røntgen af ben og knæled 31/05/2017. Fibrøs dysplasi af tibia blev afsløret. Ifølge resultaterne af en CT-scanning af underekstremiteterne dateret 1. juni 2017 blev en polyostotisk form for fibrøs dysplasi af knoglerne i underekstremiteterne afsløret.

Det blev besluttet at foretage en differentialdiagnose mellem forskellige former for polyostotisk fibrøs dysplasi. Ved opfølgningsaftalen blev detaljer om den livshistorie, som moderen tidligere havde skjult, afsløret. Uddragene indgik ikke i ambulatoriet og blev opbevaret i moderens hjem.
Fortsættelse af sygehistorien og sygdom: i en alder af seks måneder blev isoleret lache noteret. Ved 2 års alderen var der bilateral lache, brystklumper til højre med en diameter på 3,5 cm, til venstre 3 cm. Kønsformlen Ma2P0Ax0. Over 12 måneder blev brystforstørrelsen gentaget 5 gange, varigheden af fremskridt var 3 uger og regression var 2 uger. Konsulteret og undersøgt af en endokrinolog ved Instituttet for Pædiatrisk Endokrinologi i den føderale statsbudgetinstitution "National Medical Research Center of Endocrinology" i det russiske sundhedsministerium. I en alder af 4 begyndte pigen at opleve menstruationslignende udflåd. Ultralyd afslørede en follikulær cyste i højre æggestok. En operation blev udført: fjernelse af en follikulær cyste (cystektomi). Efter kirurgisk behandling forsvandt tegn på for tidlig pubertet, væksthastigheder var normale, og hævelse af mælkekirtlerne blev ikke længere observeret.
Efter at have analyseret en komplet livshistorie (herunder tidlig seksuel udvikling), samt røntgen- og computertomografidata, var der mistanke om MRD-syndrom. Pigen blev sendt til Institut for Pædiatrisk Endokrinologi, hvor hun tidligere var blevet behandlet.
Data fra laboratorie- og funktionsundersøgelser.
Klinisk analyse af blod og urin: normal.
Biokemisk blodprøve: total calcium - 2,27 (normalt 2,02-2,6 mmol/l), ioniseret calcium - 1,1 mmol/l (normalt 1,13-1,32 mmol/l), uorganisk fosfor - 3,66 mmol/l (normalt 0,86-1,56 mmol) /l), er andre indikatorer inden for normale grænser.
Biokemisk urinanalyse: normal.
Skjoldbruskkirtelprofil: TSH 3,2 µIU/l (normalt 0,32-5 µIU/l); T4 i alt 65 nmol/l (normalt 39-155 nmol/l); Fri T4 21nmol/l (normalt 13-30nmol/l).
Hormonel profil: prolaktin 6,1 ng/ml (normalt 4-23 ng/ml); somatomedinS 423ng/ml (normal 95-473ng/ml); parathyreoideahormon 16ng/l (normalt 8-24ng/ml); østradiol 24pg/ml (normalt 22-30pg/ml); FSH 6,8 mIU/l (aldersnorm 1,5-4,0 mIU/l); LH 1,75 mU/ml (aldersnorm 0,5–4,6 mU/l).
Scintigrafi med technetium: der er en stigning i akkumuleringen af det farmakologiske lægemiddel i området af den øverste tredjedel af højre lårben (102%), i området af den midterste tredjedel af højre skinneben (180%) og den højre talus (106%).
Røntgen af kraniet i lateral projektion - uden patologi.
Ultralyd af skjoldbruskkirtlen: samlet volumen 6,6 cm 3 ; ekkogeniciteten er gennemsnitlig; strukturen af parenkymet er heterogene, udvidede follikler. Ultralyd af andre indre organer var ikke bemærkelsesværdig.
Instrumentelle og laboratoriemæssige forskningsmetoder udelukkede sygdomme i biskjoldbruskkirtlerne, hypofysen, pinealkirtlen og binyrerne. Diagnosen blev bekræftet: "MRD-syndrom".
Etiotropisk behandling for polyostotisk fibrøs osteodysplasi er endnu ikke udviklet.
Anbefalet: blid ortopædisk kur, stop med at danse, bære ortopædiske indlægssåler, undgå fysisk aktivitet og skader, fysioterapi for at styrke musklerne i underekstremiteterne, lænden, ryggen, svømning. Lægemiddelbehandling blev ordineret: alfacalcidol 0,25 mcg en gang dagligt, under kontrol af biokemisk analyse, et anti-osteoporotisk middel (osteogenon 1 tablet 2 gange dagligt i 1 måned 2 gange om året), en fosfor-calcium metabolismeregulator (calcemin Advance) 1 tablet 2 gange om dagen i 1 måned 4 gange om året).
1 år senere, som følge af en krænkelse af regimet, modtog pigen et lukket brud på den midterste phalanx på den 4. tå af hendes højre fod ved trampolincentret. Røntgenbilleder af højre fod i direkte og skrå projektioner viser en patologisk fraktur af den midterste phalanx i 4. tå på højre fod, udtynding af det kortikale lag af 2. og 4. tå, og knoglestrålerne er dårligt differentierede. Immobilisering blev udført med gips i en periode på 4 uger. Efter denne periode viser røntgenbilleder i direkte og skrå projektioner diastase mellem fragmenter og forsinket konsolidering, hvilket er karakteristisk for MRD-syndrom.


Fig. 3a, b Røntgenbilleder af højre fod: a- før immobilisering, b- efter 4 ugers immobilisering
Hospitalsindlæggelse er planlagt for at justere dosis af bisfosfonater. I øjeblikket er kirurgisk behandling ikke tilrådelig, da det med begyndelsen af puberteten er muligt at bremse eller endda stoppe den videre udvikling af osteodysplasi. Muligheden for operation bør overvejes for cystiske formationer, der direkte truer en patologisk fraktur.
1. Ved mistanke om MRD-syndrom er en grundig undersøgelse af sygehistorien nødvendig, da manglen på fuldstændige data i den medicinske dokumentation ikke er ualmindeligt.
2. Diagnostiske kriterier: hudmanifestationer - café-au-lait nevi, for tidlig seksuel udvikling, fibrøs osteodysplasi. Da ufuldstændige og slettede former for MRD-syndrom er meget mere almindelige, bør det mistænkes, hvis kun to tegn er til stede.
3. Laboratorieundersøgelser (serumhormoner, biokemisk blodprøve) kan være uden patologiske ændringer. Der kan heller ikke være nogen abnormiteter i binyrerne, hypofysen, tegn på øget længdevækst, akromegali mv.
4. FSH:LH-forholdet >2,5 (i dette tilfælde 3,9) indikerer sandsynligheden for gentagelse af en ovariecyste og behovet for observation af en endokrinolog.
5. Periodisk røntgenundersøgelse er nødvendig for at påvise progression af fibrøse cyster.
Liste over forkortelser.
ACTH er adrenokortikotropt hormon.
Axe - hårvækst i armhulerne.
CT - computertomografi.
LH er luteiniserende hormon.
Træningsterapi - terapeutisk fysisk kultur.
Ma - mælkekirtler.
MRD syndrom - McCune-Albright-Braitsev syndrom.
ARVI er en akut respiratorisk virusinfektion.
PTH - parathyreoideahormon
P - kønsbehåring.
T4 - thyroxin.
TSH er skjoldbruskkirtelstimulerende hormon.
Ultralyd - ultralydsundersøgelse.
FP er et farmakologisk lægemiddel.
FSH er follikelstimulerende hormon.
cAMP - cyklisk adenosinmonofosfat.
Litteratur.
1. Makazan N.V. Rollen af post-receptor signaleringsforstyrrelser i udviklingen af multihormonal resistens og autonom hyperfunktion af de endokrine kirtler hos børn." – Diss... cand. honning. Sciences. – M., 2017.
2. Uvarova E.V. Begrundelse for brugen af urtemedicin baseret på Vitex agnus castus hos piger med tidlig pubertet forbundet med McCune-Albright-Braitsev syndrom // Pediatric and Adolescent Reproductive Health - 2017. - Nr. 6. - S.77–83.
3. Plaksina M.I., Vitebskaya A.V. Variabilitet af symptomer i McCune-Albright-Braitsev syndrom // Pædiatri - 2016. - Nr. 6 (123).
4. Albright syndrom: årsager, tegn, diagnose, hvordan man behandler // Syndrome.info: Alt om syndromer. - http://sindrom.info/olbrajta/
Albrights syndrom, eller på anden måde pseudohypoparathyroidisme, er en meget sjælden genetisk betinget sygdom, der viser sig ved forstyrrelser i calcium- og fosformetabolismen og som følge heraf patologi i skeletsystemet. Som regel er hovedsymptomerne også ledsaget af udviklingsforsinkelser - både psykiske og fysiske. Sygdommen opstår oftere hos kvinder.
Ætiologi
Forskere mener, at Albrights syndrom er forårsaget af intakte nyre- og knogleceller til parathyreoideahormoner. Dette skyldes en receptordefekt. I nyrerne forstyrres synteseprocessen af cAMP (cyklisk aminazinmonofosfat), som er et hjælpeled i de metaboliske processer forbundet med parathyreoideahormon. Sådanne skader opstår, når specifikke gener er beskadiget.
Lidelsen kan forekomme i enhver del af den metaboliske kæde. Hos nogle patienter er selve receptoren beskadiget, hos andre undergår cellemembranproteiner ændringer, hos andre er sygdommen forbundet med utilstrækkelig produktion af cAMP. Alle disse tilfælde er forbundet med den genetiske årsag til patologien.
Patogenese
Albrights syndrom er tæt forbundet med fysiologien af biskjoldbruskkirtlerne. Normalt regulerer de niveauet af calcium og fosfor i blodet, men i denne patologiske tilstand er de ligeglade med udsving i disse elementer, hvilket kan føre til sekundær hyperparathyroidisme. Men selv høje niveauer af parahormon stimulerer ikke den accelererede eliminering af fosfor og cAMP fra blodet (da nyrerne er intakte for dets virkninger) og fremkalder samtidig ændringer i knoglestrukturen.
Som regel er der en kompenserende stigning i biskjoldbruskkirtlerne, porøsitet af knoglerne, udseendet af cystiske formationer i dem, forkalkninger i huden, fedtvæv, muskler, hjerte, blodkar og bindehinden og hornhinden.
Klinik

Albrights syndrom manifesterer sig næsten på samme måde som ægte hypoparathyroidisme. Patienterne oplever kramper, som kan opstå enten af sig selv eller efter eksponering for irriterende faktorer (nervespændinger, temperaturændringer, tungt fysisk arbejde osv.). Forkalkninger i huden og subkutant fedt kan give sår og forårsage ubehag for patienterne. I dette tilfælde vil patienten søge hjælp ikke fra en endokrinolog eller genetiker, men fra en kirurg eller hudlæge.
Sådanne mennesker har tendens til at være korte, have et rundt, hævet ansigt, fedme og forkortede fingre. Pseudarthrose observeres, hvor de ikke burde være, der er mangel på mobilitet på de steder, hvor det anatomisk forventes, og en krænkelse af konfigurationen af led og knogler. Symptomerne omfatter også opkastning, blod i urinen, grå stær og ændringer i tandemaljen.
På grund af lave niveauer af calcium i blodet fra barndommen oplever patienterne desuden et fald i intelligens og udviklingsforsinkelser.
Diagnostik

Albright syndrom er en sygdom, der er ret svær at diagnosticere hos et spædbarn, men i en alder af 5-10 år opstår der ret slående manifestationer, der ikke efterlader lægen i tvivl om diagnosen. Børn har flere misdannelser i knoglesystemet. Niveauet af calcium i blodet er reduceret, og niveauet af fosfor og parathyreoideahormon øges.
For at bekræfte diagnosen kan du teste mængden af fosfat og cAMP i urinen. For at gøre dette får patienten 200 enheder parathyreoideahormon intravenøst, og derefter opsamles en portion urin efter 4 timer. Hvis en signifikant stigning ikke observeres, kan dette indikere resistens i nyrevævet over for parathyreoideahormon.
Disse tegn er nok til at diagnosticere Albright syndrom. Diagnostik kan suppleres med røntgen. Det gør det muligt at se specifikke ændringer i patientens skeletsystem og bløde væv.
Ud over parathyreoideahormon har personer med denne diagnose også resistens over for andre hormoner, så kvinder er differentielt diagnosticeret med Shereshevsky-Turner syndrom. Disse to sygdomme har eksterne ligheder, og for at verificere diagnosen skal lægen ordinere en test for kønskromatin og give en henvisning til en gynækologisk undersøgelse og ultralyd af bækkenorganerne.
Behandling og prognose

Når en diagnose af Albright syndrom er stillet, skal behandlingen påbegyndes uden forsinkelse, da forsinkelser forværrer mentale udviklingsproblemer. Børn får ordineret calciumtilskud i tilstrækkelige daglige doser til at opretholde normale serumkoncentrationer. Derefter tilføjer de D-vitamin i en dosis, der ikke overstiger 100.000 enheder om dagen.
For at justere behandlingen er det nødvendigt at overvåge calciumniveauer hver 7. dag i de første to uger og derefter en gang om måneden indtil slutningen af behandlingen. Når den optimale koncentration af calcium i blodet er opnået, kan du tjekke den en gang i kvartalet.
For at eliminere symptomerne på sekundær hyperparathyroidisme anbefales det at følge en diæt med en reduceret mængde fosfor. Hvis en mangel på andre hormoner opdages, udføres erstatning og symptomatisk behandling.
Hvis behandlingen påbegyndes til tiden, og behandlingen er rationel, er prognosen for livet gunstig. Men før graviditeten skal kvinder med denne diagnose gennemgå konsultation med en genetiker.
Albright syndrom er en sjælden genetisk lidelse af ukendt oprindelse. Det er arveligt og er hovedsageligt kendetegnet ved skader på skeletsystemet. Findes oftest hos hunner. Men der er tidspunkter, hvor drenge også kan blive syge. De fleste patienter med dette syndrom lider af fysisk og mental retardering.
Sygdommen blev opkaldt efter den videnskabsmand, der opdagede den. Skeletsystemet begynder at nedbrydes under påvirkning af calcium- og fosforstofskiftet. Biskjoldbruskkirtlen udskiller biskjoldbruskkirtlen hos en person med Albright syndrom, vævsresistens over for parathyreoideahormon er svækket.
Symptomer på Butler Albrights syndrom
Der er tre hovedtræk sygdomme. Hvis selv to ud af tre er til stede, kan en sådan diagnose stilles.
- Meget tidlig pubertet som følge af højt forhøjede hormonniveauer. Piger kan begynde menstruation i de første måneder efter fødslen. Drenge med Albrights syndrom oplever meget sjældent tidlig pubertet. Ændringer i hormonelle niveauer opstår på grund af ukorrekt funktion af binyrerne, hypofysen og skjoldbruskkirtlen. Dette symptom kan forekomme i komplet eller ufuldstændig form. I fuld form begynder menstruationen meget tidligt og er normalt tung og smertefuld. I spædbarnet manifesterer dette syndrom også sekundære seksuelle egenskaber, for eksempel hævelse af mælkekirtlerne. Hvis manifestationen er ufuldstændig, forekommer menstruation ikke. Meget ofte, med Albright syndrom, udvikler piger cyster på æggestokkene. Hos drenge observeres i tidlig pubertet penisforstørrelse og kønsbehåring.
- Fibrøs osteodysplasi. Denne sygdom indikerer en krænkelse af knoglevæv, hvilket fører til krumning og deformation af knogler. Karakteriseret af asymmetri. De mest almindelige tegn er halthed og krumning af rygsøjlen. Lange knogler er oftest påvirket af ændringer. Skeletvæksten bremses, dette er mærkbart selv hos børn. Mange ofre for McCunes syndrom oplever ujævn vækst af kranieknoglerne, hvilket fører til ekstern deformitet. Når ansigtet ændrer sig, kan der opstå svulmende øjne. Efter pubertetsalderen bremses knogleforandringer, og forekomsten af brud falder.
- Ændringer i huden. Viser sig i øget pigmentering. Huden på torso og lår bliver dækket af gulbrune pletter med takkede kanter. Pigmentering kan ses på lænden, øvre ryg, balder og bryst. Pletterne vises umiddelbart efter barnet er født.

Behandling af Albright (McCune) syndrom
 Patienter, der lider af McCune syndrom, skal til enhver tid være under opsyn af specialiserede specialister (gynækolog, endokrinolog, traumatolog, øjenlæge).
Patienter, der lider af McCune syndrom, skal til enhver tid være under opsyn af specialiserede specialister (gynækolog, endokrinolog, traumatolog, øjenlæge).
Kompleks behandling er påkrævet. Da sygdommen er karakteriseret ved en genetisk mutation, er det først og fremmest nødvendigt at slippe af med symptomerne på dens manifestation.
Obligatorisk hormonbehandling er indiceret at vedligeholde det endokrine system.
Det er nødvendigt ikke at gå glip af det øjeblik, hvor knoglerne i ansigtet begynder at deformeres for at forhindre disse ændringer under sygdom.
Nogle gange er det påkrævet kirurgisk indgreb. Det er ordineret, når der på grund af ændringer i kraniets struktur er risiko for høre- eller synstab.
Ved smerter anvendes specielle smertestillende midler indeholdende små mængder bisfosfonater.
Det er meget vigtigt at træffe forebyggende foranstaltninger for at styrke musklerne.
For hver patient, der lider af Albrights sygdom, vælger lægen sin egen individuelle behandling, som skal følges nøje.
Skal konstant overvåges mængden af calcium i kroppen. Ved start af terapi udføres sådanne undersøgelser en gang om ugen, hvorefter deres frekvens reduceres til en gang om måneden, indtil hovedbehandlingen er afsluttet.
Under behandlingen er det meget vigtigt at følge en diæt, hvor mængden af fødevarer, der indeholder fosfor, er reduceret til et minimum.
Det skal bemærkes, at en kvinde, der lider af dette syndrom Når du beslutter dig for at blive gravid, bør du helt sikkert rådføre dig med en god genetiker.
Albrights sygdom (McCune) forekommer hos én person ud af 1.000.000. Specifik behandling for dette syndrom er endnu ikke opfundet, men man bør ikke miste håbet. Medicin står ikke stille, og læger vil hjælpe med at overvinde ubehagelige øjeblikke og vil holde patienten under observation til enhver tid.
Catad_tema Pædiatri - artikler
Pseudohypoparathyroidisme (Albrights arvelige osteodystrofi): vanskeligheder med differentialdiagnostisk søgning. Klinisk observation
E.V. Tozliyan, pædiatrisk endokrinolog, genetiker, Ph.D., I.V. Shulyakova, neurolog, Ph.D.,
separat strukturel enhed "Research Clinical Institute of Pediatrics" af den statsbudgettære uddannelsesinstitution for videregående professionel uddannelse "Russian National Research Medical University opkaldt efter N.I. Pirogov" fra Sundhedsministeriet i Den Russiske Føderation, Moskva
Nøgleord:
børn, pseudohypoparathyroidisme, Albrights arvelige osteodystrofi, fedme, hypocalcæmi, diagnose, resistens over for parathyreoideahormon.
Nøgleord: børn, pseudohypoparathyroidisme, Albright arvelig osteodystrofi, fedme, hypocalcæmi, diagnostik, parathyreoideahormonresistens.
Pseudohypoparathyroidisme (græske pseuder - falsk + hypoparathyroidisme; synonym: Albrights arvelige osteodystrofi, javanesisk kyllingesyndrom) er en sjælden arvelig sygdom i skeletsystemet, der simulerer hypoparathyroidisme og er karakteriseret ved nedsat calcium- og fosformetabolisme; ofte ledsaget af forsinket mental og fysisk udvikling. Sygdommen blev første gang beskrevet af den amerikanske endokrinolog Albright F. i 1942. Forekomsten af sygdommen er 7,9 pr. 1 million mennesker.
GENETISKE DATA
Pseudohypoparathyroidisme (PHP) er en genetisk heterogen sygdom. Data om typen af arvelig overførsel er modstridende: både X-bundne dominante og autosomale dominante, autosomale recessive typer. I de fleste tilfælde er udviklingen af Albrights arvelige osteodystrofi forbundet med mutationer i 20q13-locuset af GNAS1-genet placeret på kromosom 20 (Patten et al., 1990), der koder for Gs-alfa-proteinet forbundet med parathyroidhormon (PTH)-receptoren . En lignende fænotype blev også påvist hos patienter med interstitiel deletion af den lange arm af kromosom 2 ved 2q37-locuset.
PATOGENESE
Patogenesen af pseudohypoparathyroidisme er baseret på genetisk bestemt resistens i nyrerne og skelettet mod virkningen af parathyreoideahormon som følge af en defekt i det "specifikke cytoreceptor - parathyroidhormon - adenylatcyklase", som forstyrrer dannelsen af cyklisk 3"- , 5"-adenosinmonofosfat (cAMP) i nyrerne, som er intracellulær mediator af biskjoldbruskkirtlens virkning på metaboliske processer. Pseudohypoparathyroidisme er genetisk heterogen sygdom. Hos nogle patienter er selve cytoreceptoren, der binder parathyreoideahormon, defekt (type 1A pseudohypoparathyroidisme, andre har en defekt i det nukleotidbindende protein, lokaliseret i lipiddobbeltlaget i cellemembranen og binder receptoren til adenylatcyklase (type 1B); pseudohypoparathyroidisme). Nogle patienter har en enzymatisk mangel på selve adenylatcyclase (type 2 pseudohypoparathyroidisme). cAMP-manglen som følge af disse defekter fører til forstyrrelse af syntesen af specifikke proteiner, der bestemmer den biologiske virkning af parathyreoideahormon. Således går målorganernes følsomhed over for parathyreoideahormon tabt.
KLINISKE EGENSKABER
I øjeblikket er der 4 kliniske former for patologi: type 1A, 1B, 1C og 2. Kendskab til deres kliniske og biokemiske egenskaber og genetiske forskningsdata giver mulighed for differentialdiagnose inden for selve den nosologiske form.
Almindelige tegn, der tillader en mistanke om sygdommen, er disproportionalitet i fysisk udvikling, kort statur (til punktet af dværgvækst) på grund af afkortning af underekstremiteterne (foto 1), brachydactyly (foto 2) og en rund "måneformet" ansigt (foto 3). Exostoser og dental aplasi observeres nogle gange.
Foto 1.
Udseende af et barn med Albrights osteodystrofi
(træk ved fænotypen, kort statur på grund af afkortning af underekstremiteterne)
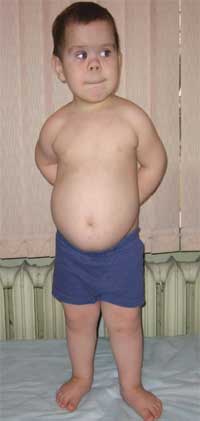
Foto 2.
Funktioner af patientens skeletsystem
med Albrights osteodystrofi
(brachydactyly - forkortelse af fingre)
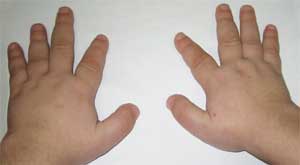
Foto 3.
Funktioner af barnets fænotype
med Albrights osteodystrofi
(rundt måneformet ansigt)

Et patognomonisk tegn er en skarp afkortning af I, III og V metacarpal og metatarsale knogler (især III og IV), som resulterer i, at II fingre på hænder og fødder er længere end de andre, og når hånden er knyttet sammen ind i en knytnæve, er der ingen buler i området af IV og V metacarpophalangeal leddene - såkaldt brachymetaphalangisme. Korte brede phalanges, fortykkelse af kraniehvælvingen og demineralisering af knogler (osteoporose) og fedme påvises også.
Mental retardering (normalt moderat sværhedsgrad) findes hos ca. 20 % af patienterne. Ifølge nogle forfattere forekommer mental retardering i 70% af tilfældene i nærvær af hypocalcæmi og i 30% af tilfældene i normocalcæmi. Psykiske processer hos patienter bremses. I den neurologiske status bemærkes ofte motorisk kejtethed og neurotiske reaktioner: frygt, angst, rastløshed, dårlig søvn, øgede reflekser, kramper, der er tetaniske i naturen og forårsaget af hypocalcæmi, og nogle gange krampagtige paroxysmer. Myopatiske symptomer er også blevet beskrevet: muskeltræthed, muskelsvaghed. Ekstrapyramidale lidelser observeres ofte: choreiform hyperkinesis, athetose, ansigtshemispasme, parkinsonisme i nogle tilfælde forekommer epileptiske paroxysmer, cerebellare symptomer: ataksi, tab af koordination.
Forkalkning af blødt væv og subkutane forkalkninger (bryst, mave, hælsener) påvises ofte, og histologisk undersøgelse af dem viser osteoma cutis(Izraeli et al., 1992), hjerne (basale ganglier). Det er vigtigt at bemærke, at forkalkninger kan være til stede allerede ved fødslen. Som et resultat af hypocalcæmi udvikles grå stær normalt, og der opstår tandemaljefejl.
PSEUDOHYPOPARATROOSE TYPE 1A
har et autosomalt dominant arvemønster. Genet for pseudohypoparathyroidisme type 1A - GNAS1 - er lokaliseret på den lange arm af kromosom 20, i 20q13.2 locus. Udviklingen af sygdommen er forbundet med en mangel på guanin-nukleotid-bindende protein (Gs-protein). Samtidig er PTH, der binder til receptorer i målvæv, ikke i stand til at aktivere cyklisk adenosinmonophosphat (cAMP) og forårsage et vævsrespons. Sandsynligvis ligger en lignende mekanisme til grund for udviklingen af ufølsomhed af væv i andre organer og endokrine kirtler (hypofunktion af skjoldbruskkirtlen, kønskirtler, hypofysen, diabetes mellitus samt en reduceret respons fra leveren på glukagonadministration), observeret ved pseudohypoparathyroidisme type 1A. I denne type patologi observeres den normale øgede udskillelse af cAMP i urinen som reaktion på eksogen administration af PTH ikke. Sygdommen diagnosticeres oftere i en alder af 5-10 år. Patienter har kort statur, en kort hals, et rundt ansigt, afkortning af metacarpal og metatarsal knogler (normalt afkortning af den fjerde og sjældnere den anden fingre) - den såkaldte brachy-metaphalangisme. Forkalkning af blødt væv og subkutane forkalkninger noteres, som kan påvises ved fødslen; Samtidig involvering af andre endokrine kirtler observeres ofte: skjoldbruskkirtlen (hypofunktion), kønskirtler, bugspytkirtel (diabetes mellitus). Som følge af hypocalcæmi udvikles ofte grå stær og tandemaljefejl. Som en differentialdiagnostisk test til at skelne PHP type 1A fra hypoparathyroidisme: fraværet af en klinisk effekt fra parenteral administration af PTH i form af en stigning i niveauet af calcium i blodet og en stigning i den renale udskillelse af fosfor i urinen (fosfatureffekt).
Biokemisk undersøgelse afslører hypocalcæmi, hyperfosfatæmi, øgede niveauer af parathyreoideahormon i blodet og hypofosfaturi. Niveauet af Gs-protein i blodet reduceres. Røntgenundersøgelse af skeletsystemet afslører afkortning af metacarpal og metatarsale knogler, generaliseret demineralisering og fortykkelse af knoglerne i kraniehvælvingen.
PSEUDOHYPOPARATROOSE TYPE 1B
har en autosomal dominant arvetype, men en dominant, X-bundet arvetype er ikke udelukket. Det er nødvendigt at huske på den nogle gange observerede ufuldstændige penetrering af sygdomsgenet og muligheden for latent transport af patologien. Derfor anbefales klinisk (påvisning af subklinisk sygdomsforløb) og biokemisk undersøgelse (bestemmelse af calcium-, fosfor-, PTH-niveauer i blodet) af mistænkte bærere af sygdommen. PHP type 1B er forårsaget af en mangel på vævsreceptorer for parathyreoideahormon i målorganer og begrænset resistens over for parathyreoideahormon. Det kliniske billede ligner det for type 1A, men der er ingen skade på andre endokrine kirtler, og osteodystrofi er mindre almindelig.
Patienter har ikke en nyrerespons på eksogen administration af parathyroidhormon i form af en stigning i udskillelsen af cyklisk adenosinmonofosfat i urinen, men i modsætning til type 1A er niveauet af Gs-protein i blodet normalt. Kvinder rammes oftere end mænd, men sværhedsgraden af sygdommen kan være den samme hos både mænd og kvinder.
PSEUDOHYPOPARATROOSE TYPE 1C
nogle forfattere identificerer det med pseudo-pseudohypoparathyroidisme (PPHP), beskrevet af Albright F. i 1952. Det er karakteriseret ved et klinisk billede, der er karakteristisk for PHP, men niveauerne af calcium og fosfor i blodet og urinen forbliver inden for normale grænser. Niveauerne af PTH- og Gs-protein i blodet forbliver også på normale niveauer. Nogle patienter med PHP type 1C har sletninger de novo på kromosom 2. Det er muligt, at denne variant af sygdommen er en undertype af PGP type 1A.
PSEUDOHYPOPARATROOSE TYPE 2
klinisk ligner andre typer af sygdommen, men har en autosomal recessiv arvemåde. Eksistensen af autosomalt dominerende former for patologi kan ikke udelukkes. Udviklingspatogenese er forbundet med intracellulær resistens over for cAMP. PTH binder sig derefter til receptorer og forårsager en normal cellulær respons på PTH i form af øget cAMP-udskillelse. Intracellulær ufølsomhed over for cAMP tillader imidlertid ikke den fulde effekt af PTH at blive realiseret. Samtidig opretholdes nyrernes normale reaktion på eksogen administration af parathyreoideahormon i form af øget udskillelse af cyklisk adenosinmonofosfat i urinen. Det er blevet foreslået, at PHP type 2 kan være forbundet med D-vitaminmangel.
De identificerede typer af PGP er således klinisk karakteriseret ved reduceret følsomhed af målorganer over for PTH, men adskiller sig i de patogenetiske mekanismer for dannelsen af vævsufølsomhed.
DIAGNOSTIK
En differentialdiagnostisk laboratorietest kan være mønsteret for renal udskillelse af cAMP som reaktion på PTH-administration: øget udskillelse af cAMP observeres i type 2 og fravær af cAMP i type 1. Diagnosen bekræftes ved påvisning af et reduceret niveau af guaninnukleotid bindende protein (Gs-protein) i blodet (i gennemsnit 1,5-2 gange) i forhold til normen. Hypocalcæmi er normalt kombineret med hyperphosphatæmi og hypophosphaturi. PTH-niveauer er forhøjede; i type 1C er PTH-niveauet normalt, hvilket giver anledning til navnet "pseudohypoparathyroidisme". Røntgenundersøgelse af skeletsystemet afslører afkortning af metacarpal og metatarsale knogler, ofte generaliseret demineralisering (osteoporose) og fortykkelse af knoglerne i kraniehvælvingen. Det dermatoglyfiske mønster viser en forskydning af den aksiale palmar triradius.
Diagnosekriterier:
- kort statur;
- rundt ansigt;
- forsinket neuropsykisk udvikling;
- skelet abnormiteter;
- lavt serum calcium;
- højt niveau af parathyroidhormon i blodet;
- nedsat urinudskillelse af fosfater og cAMP.
BEHANDLING OG FOREBYGGELSE
Behandling af hypocalcæmi består i at ordinere calciumtilskud i doser, der er tilstrækkelige til at opretholde normale calciumkoncentrationer i blodet. D-vitaminbehandling er af stor betydning I øjeblikket anvendes aktive metabolitter af D-vitamin - oxidevit, 1-alfa-D3, calcitrin osv. i en dosis på 1-2 mcg/dag med positive resultater (forhøjede calciumniveauer i blodet. , nedsatte manifestationer af konvulsivt syndrom). Tachistin (0,5-1,5 mg/dag) er også effektivt. Dette lægemiddel øger optagelsen af calcium i tarmene og hjælper derved med at øge niveauet af calcium i blodet. Antikonvulsiv terapi anvendes som supplerende behandling. Behandlingen har ikke en mærkbar effekt på intellektuel udvikling, men sammen med et fald i symptomerne på konvulsivt syndrom observeres regression af neurologiske manifestationer (subkortikale lidelser, choreiform hyperkinesis, athetose osv.). For at undgå overdosering af D-vitaminpræparater er det nødvendigt at overvåge koncentrationen af calcium i blodet hver 3-7 dag i de første 2 uger af behandlingen og hver måned i de næste 2-3 måneder. Når en stabil calciumkoncentration i blodet er nået, er det nok at kontrollere den en gang hver 2-3 måned. En phosphor-begrænset diæt hjælper med at normalisere både fosfor- og calciumkoncentrationer i blodet og eliminere symptomerne på sekundær hyperparathyroidisme. I tilfælde af insufficiens af andre endokrine kirtler udføres erstatningsterapi med passende hormoner.
Behandling med parathyreoideahormon er ineffektiv. For at lindre krampeanfald indgives en 10% opløsning af calciumchlorid eller calciumgluconat intravenøst; oralt – 5-10% opløsning af calciumchlorid, 1 spiseskefuld 3-4 gange om dagen: calciumgluconat, calciumlactat – op til 10 g pr. dag.
VEJRUDSIGT for livet er bestemt af sværhedsgraden af det konvulsive syndrom.
FOREBYGGELSE sygdom er baseret på data fra medicinsk genetisk rådgivning.
MEDICINSK-GENETISK RÅDGIVNING
Når man udfører medicinsk genetisk rådgivning, bør man tage udgangspunkt i den autosomalt dominerende arvetype og den høje (50 %) risiko for tilbagefald af sygdommen i familien med arvelige former. For at identificere arten af arvetypen er det nødvendigt at foretage en grundig undersøgelse af forældrene, da syndromet kan manifestere sig med minimale kliniske symptomer. I øjeblikket er molekylærgenetisk diagnosticering af sygdommen blevet udviklet og forbedres ved at typebestemme mutationer i GNAS1-genet på kromosom 20. Metoder til prænatal diagnosticering af sygdommen generelt og dens individuelle typer er under udvikling.
KLINISK OBSERVATION Drengen G., 14,5 år gammel (foto 4), blev indlagt på Research Clinical Institute of Pediatrics med diagnosen degenerativ sygdom i nervesystemet? medfødt ekstern hydrocephalus; symptomatisk epilepsi; arveligt syndrom? opbevaringssygdom? metabolisk encefalopati; subklinisk hypothyroidisme; kort statur af blandet oprindelse; kognitiv svækkelse.
Klager ved indlæggelse af intens paroxysmal hovedpine lokaliseret i frontalregionen og ledsaget af opkastninger, hvilket bringer lindring, nedsat hukommelse og præstation i skolen, krampeanfald, hvor der opstår trækninger i højre hånd.
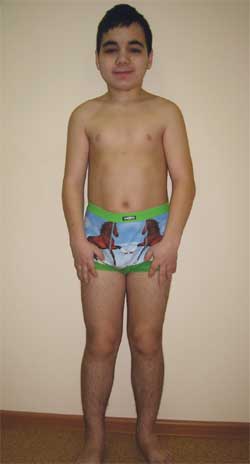
Foto 4.
Barn G., 14,5 år gammel, med Albrights osteodystrofi
(fænotypetræk, kort statur, forkortede lemmer, brachydactyly)
Familie historie: forældre er armeniere efter nationalitet, ikke beslægtet af blod og har ingen erhvervsmæssige farer. Der var ingen tilfælde af psykisk sygdom, epilepsi eller udviklingsforsinkelser i stamtavlen. Søskende, søster, 17 år, siges at være rask.
Historie om liv og sygdom: dreng fra 2. graviditet, som forløb uden nogen ejendommeligheder, anden fødsel, ved termin, fysiologisk, fødselsvægt - 3100 g, længde - 51 cm Han råbte med det samme, Apgar score - 7/9 point. Forværring af tilstanden på 3. dag – neonatale kramper, stoppet på barselshospitalet. Den tidlige postnatale periode er uden træk. Der var en lille forsinkelse i motorisk udvikling i det første leveår, selvstændig gang fra 1 år 3 måneder. I forbindelse hermed blev han observeret af en neurolog med diagnosen organisk skade på centralnervesystemet; medfødt hydrocephalus; neonatale anfald; historie med feberkramper.
Jeg fik diacarb og finlepsin. Start af anfald ved 1 år 11 måneder. – asymmetrisk, styrkende i form af spændinger i højre arm og ben, med rullende øjne, op til 2 minutter, uden tab af bevidsthed, hyppigt op til 10 episoder om dagen. Jeg modtog Depakine uregelmæssigt. På baggrund af uafhængig tilbagetrækning - en enkelt tonic status. Ved 2 års alderen blev der foretaget en CT-scanning af hjernen på bopælen, hvor enkelte demyeliniseringsfoci blev identificeret i occipitallapperne.
En neurokirurg blev konsulteret, og konservativ behandling blev anbefalet. Fra 3 års alderen er der en forsinkelse i psyko-tale udvikling, og observation af en psykiater anbefales.
Fra 4-5 års alderen begyndte forældre at bemærke deformation og afkortning af fingre og tæer, især 2.-4. fingre symmetrisk på arme og ben, og et fald i vækstindikatorer. Ved 8-års alderen anbefales logopædens konklusion en generel taleforstyrrelse på 2.-3. niveau. I samme alder undersøgelse af en genetiker på bopælen, konklusion: arvelig stofskiftesygdom? En undersøgelse af blodaminosyrer blev anbefalet, ingen ændringer blev opdaget; endelig konklusion: ingen tegn på en arvelig metabolisk sygdom blev identificeret; hypokondroplasi; Behandling hos neurolog og endokrinolog anbefales.
Bord.
Profil for mental udvikling af barn G., 14,5 år gammel (IQ = 68)

I en alder af 8 år blev han konsulteret af en endokrinolog vedrørende forsinket vækst og udvikling. En røntgenundersøgelse af hænderne afslørede følgende træk: mellem- og hovedfalangerne og metakarpale knogler blev forkortet og fortykket; Radiologens diagnose var achondroplasi.
Gentagne gange undersøgt på bopælen på et neurologisk hospital. I en alder af 12 forekom krampeanfald uden bevidsthedstab med trækninger i højre arm, som var af serielle karakter. Antikonvulsiv terapi (Depakine) blev ordineret, og anfaldshyppigheden faldt betydeligt. I en alder af 13 blev der foretaget en MR af hjernen med kontrast - symmetriske ændringer i tindingelappernes basis i niveau med kernerne i form af en stigning i MR-signalet, som er typisk for toksisk (mangan) eller metaboliske (kobber, jern) encephalopatier.
Reeksamineret i en alder af 13 år 3 måneder. en endokrinolog, en undersøgelse af skjoldbruskkirtelprofilen afslørede en stigning i thyreoidea-stimulerende hormon (TSH), subklinisk hypothyroidisme blev diagnosticeret, og L-thyroxin blev ordineret.
Ved analyse af barnets ambulatoriekort og dokumentation på bopælen er der én gang foretaget kalcium- og fosforundersøgelse, i en alder af 1,5 år konstateret hypocalcæmi, men der er ikke foretaget yderligere undersøgelse ved denne lejlighed. I betragtning af usikkerheden ved diagnosen på bopælsstedet sendte genetikeren barnet til Moskva, til Scientific Research Clinical Institute of Pediatrics, for at afklare diagnosen.
Objektive forskningsdata:
Højde – 143 cm, vægt – 43 kg.
Fysisk udvikling er meget lav, harmonisk, fysikken er uforholdsmæssig på grund af afkortning af lemmerne. Vækst Sds svarer til –2,8 afvigelser fra normen (norm –2+2).
Fænotypetræk: rundt ansigt, kort hals, anti-mongoloide palpebrale fissurer, bred næsebro, høj pande, brachydactyly, afkortning af IV og V metacarpal og metatarsale knogler (foto 5). Indre organer – uden nogen ejendommeligheder. Seksuel udvikling – Tanner stadium III-IV (som svarer til alder).
Laboratorie- og funktionsforskningsdata:
Klinisk analyse af blod og urin er normal.
Biokemisk blodprøve: totalt calcium – 1,39 (normalt 2,02-2,6 mmol/l), ioniseret calcium – 0,61 (normalt 1,13-1,32 mmol/l), uorganisk fosfor – 3,66 (normalt 0,86-1,56 mmol/l), andre indikatorer er inden for normale grænser.
Biokemisk urinanalyse: renal udskillelse af fosfater reduceres - 11,5 mmol/l (normalt 19-32 mmol/l).
Skjoldbruskkirtelprofil: TSH – 11,75 (normalområde 0,4–4,0 µIU/ml), fri T4 – 0,49 (normalområde 1,0–1,8 ng/dL).
Parathyreoideahormon – 499 (normalt 12-65 pg/ml), STH – 7 ng/ml (normalt 7-10 ng/ml), somatomedin-C – 250 ng/ml (normalt 88-360 ng/ml).
Ultralyd af indre organer - uden nogen funktioner.
EKG – migration af den supraventrikulære pacemaker på baggrund af en regulær hjertefrekvens på 71-80 slag/min. Ufuldstændig blokade af højre bundtgren. Afbrydelse af repolariseringsprocessen i myokardiet i den venstre ventrikels bagvæg (fald i ZT III, aVF).
R-grafi af rygsøjlen - højresidig skoliose af thoraxrygsøjlen af 1. grad, svær osteoporose.
R-grafi af hænderne med fangst af underarmene - afkortning og udvidelse af de terminale og midterste phalanges. Knoglealder – 13,5-14 år.
Ingen EEG-mønstre af epileptisk aktivitet blev registreret.
MR af hjernen - MR-billede af flere subkortikale foci af øget MR-signal i frontallapperne, ekstern kompenseret hydrocephalus med atrofi af hjernesubstansen.
MSCT af hjernen viser symmetriske områder med forkalkning af lentiforme kerner. Diffuse hypertætte områder i thalami, caudate kerner med et område med forkalkning til højre. Flere punktforkalkninger af det integumentære bløde væv i kraniet.
Audiogram - uden patologi.
DNA-diagnostik i GNAS1-genet er i gang.
Specialistkonsultationer:
Endokrinolog – Albrights arvelige osteodystrofi type 1A (pseudohypoparathyroidisme). Primær hypothyroidisme, ufuldstændig medicinkompensation.
Øjenlæge – komplet sekundær grå stær. Kirurgisk behandling anbefales.
Psykolog - kognitiv svækkelse (barnets psykologiske profil er præsenteret i tabellen).
Under hensyntagen til barnets fænotype, sygehistorie, resultater af yderligere undersøgelser (hypocalcæmi, hyperfosfatæmi, hypofosfaturi, øget parathyreoideahormon i blodet), forkalkninger i hjernen, tilstedeværelsen af grå stær, hypothyroidisme), blev der stillet en diagnose: Albrights arvelige osteodystrofi type 1A (pseudohypoparathyroidisme). Det anbefales at udføre DNA-diagnostik - søgning efter mutationer i GNAS1-genet.
Behandling: Barnet anbefales at tage eutirox i en dosis på 100 mcg/dag; aktiv metabolit af vitamin D - alfa-D3 (Teva) i en dosis på 2 mcg/dag; calcium (Sandoz) 2000 mg/dag; konstant brug af antikonvulsiv terapi - finlepsin 800 mg/dag under tilsyn af en neurolog-epileptolog; klasser med en talepædagog og psykolog; energi-tropisk terapi (Elkar og coenzym Q10 i aldersrelaterede doser). Overvågningsindikatorer for fosfor-calciummetabolisme, niveauer af parathyreoideahormoner.
Dermed, Den præsenterede kliniske observation demonstrerer kompleksiteten af den differentialdiagnostiske søgning, vigtigheden af rettidig undersøgelse af simple biokemiske parametre (i tilfælde af epilepsi kræves gentagen screening af fosfor-calciummetabolismeindikatorer), resultaterne af sen diagnose af en genetisk bestemt sygdom , behovet for at integrere individuelle tegn i den generelle fænotype af en bestemt patologisk tilstand for målrettet rettidig diagnose af visse former for arvelige sygdomme. Rettidig diagnose og afklaring af tilblivelsen af hvert syndrom er særligt vigtige, da de giver mulighed for at finde den optimale tilgang til behandling af disse tilstande og forebyggelse af mulige komplikationer (op til og med handicap af barnet); forebyggelse af tilbagefald af arvelige sygdomme i ramte familier (medicinsk og genetisk rådgivning). Dette dikterer behovet for læger af forskellige specialer for klart at navigere i strømmen af arvelig bestemt patologi. Referencelisten findes på redaktionen.



