Шоколад виды шоколада из чего состоит. Старт в науке. Теоретическое обоснование проблемы
– общее, системное заболевание организма, при котором происходит отложение специфического гликопротеида (амилоида) в органах и тканях с нарушением функции последних. При амилоидозе могут поражаться почки (нефротический синдром, отечный синдром), сердце (сердечная недостаточность, аритмии), ЖКТ, опорно-двигательный аппарат, кожа. Возможно развитие полисерозита, геморрагического синдрома, психических нарушений. Достоверной диагностике амилоидоза способствует обнаружение амилоида в биопсийных образцах пораженных тканей. Для лечения амилоидоза проводится иммунодепрессивная и симптоматическая терапия; по показаниям - перитонеальный диализ, трансплантация почек и печени.
МКБ-10
E85

Общие сведения
Амилоидоз – заболевание из группы системных диспротеинозов, протекающее с образованием и накоплением в тканях сложного белково-полисахаридного соединения - амилоида. Распространенность амилоидоза в мире в значительной мере географически детерминирована: так, периодическая болезнь чаще встречается в странах средиземноморского бассейна; амилоидная полиневропатия – в Японии, Италии, Швеции, Португалии и т. д. Средняя частота амилоидоза в популяции составляет 1 случай на 50 тыс. населения. Болезнь обычно развивается у лиц старше 50-60 лет. Учитывая тот факт, что при амилоидозе поражаются практически все системы органов, заболевание изучается различными медицинскими дисциплинами: ревматологией , урологией, кардиологией, гастроэнтерологией, неврологией и др.
Причины амилоидоза
Вопросы этиологии первичного амилоидоза до конца не изучены. Вместе с тем, известно, что вторичный амилоидоз обычно ассоциируется с хроническими инфекционными (туберкулезом , сифилисом , актиномикозом) и гнойно-воспалительными заболеваниями (остеомиелитом , бронхоэктатической болезнью , бактериальным эндокардитом и др.), реже - опухолевыми процессами (лимфогранулематозом , лейкозом , раком висцеральных органов). Реактивный амилоидоз может развиваться у больных с атеросклерозом , псориазом , ревмопатологией (ревматоидным артритом , болезнью Бехтерева), хроническим воспалением (неспецифическим язвенным колитом , болезнью Крона), мультисистемными поражениями (болезнью Уиппла , саркоидозом) . Среди факторов, способствующих развитию амилоидоза, первостепенное значение имеют гиперглобулинемия, нарушения функционирования клеточного иммунитета, генетическая предрасположенность и др.
Патогенез
Среди многочисленных версий амилоидогенеза наибольшее число сторонников имеют теория диспротеиноза, локального клеточного генеза, иммунологическая и мутационная теории. Теория локального клеточного генеза рассматривает лишь процессы, происходящие на клеточном уровне (образование фибриллярных предшественников амилоида системой макрофагов), в то время как образование и накопление амилоида происходит вне клетки. Поэтому теория локального клеточного генеза не может считаться исчерпывающей.
Согласно теории диспротеиноза, амилоид является продуктом аномального белкового обмена. Основные звенья патогенеза амилоидоза - диспротеинемия и гиперфибриногенемия способствуют накоплению в плазме грубодисперсных фракций белка и парапротеинов. Иммунологическая теория происхождения амилоидоза связывает образование амилоида с реакцией антиген-антитело, в которой антигенами выступают чужеродные белки или продукты распада собственных тканей. При этом отложение амилоида происходит преимущественно в местах формирования антител и избытка антигенов. Наиболее универсальной является мутационная теория амилоидоза, учитывающая огромное разнообразие мутагенных факторов, имеющих возможность вызвать аномальный синтез белка.
Амилоид представляет собой сложный гликопротеид, состоящий из фибриллярных и глобулярных белков, тесно связанных с полисахаридами. Амилоидные отложения накапливаются в интиме и адвентиции кровеносных сосудов, строме паренхиматозных органов, железистых структурах и т. д. При незначительных отложениях амилоида изменения выявляются лишь на микроскопическом уровне и не приводят к функциональным нарушениям. Выраженное скопление амилоида сопровождается макроскопическими изменениями пораженного органа (увеличением объема, сальным или восковым видом). В исходе амилоидоза развивается склероз стромы и атрофия паренхимы органов, их клинически значимая функциональная недостаточность.
Классификация
В соответствии с причинами различают первичный (идиопатический), вторичный (реактивный, приобретенный), наследственный (семейный, генетический) и старческий амилоидоз. Встречается различные формы наследственного амилоидоза : средиземноморская лихорадка, или периодическая болезнь (приступы жара, боли в животе, запор, диарея, плеврит, артрит, высыпания на коже), португальский нейропатический амилоидоз (периферическая полинейропатия, импотенция, нарушения сердечной проводимости), финский тип (атрофия роговицы, краниальная невропатия), датский вариант (кардиопатический амилоидоз) и мн. др.
В зависимости от преимущественного поражения органов и систем выделяют нефропатический (амилоидоз почек), кардиопатический (амилоидоз сердца), нейропатический (амилоидоз нервной системы), гепатопатический (амилоидоз печени), эпинефропатический (амилоидоз надпочечников), АРUD-амилоидоз, амилоидоз кожи и смешанный тип заболевания. Кроме этого, в международной практике принято различать локальный и генерализованный (системный) амилоидоз. К локализованным формам, как правило, развивающимся у лиц старческого возраста, относятся амилоидоз при болезни Альцгеймера, сахарном диабете 2-го типа , эндокринных опухолях, опухолях кожи, мочевого пузыря и др. В зависимости от биохимического состава амилоидных фибрилл среди системных форм амилоидоза выделяют следующие типы:
- AL - в составе фибрилл легкие цепи Ig (при болезни Вальденстрема, миеломной болезни , злокачественных лимфомах);
- AA – в составе фибрилл острофазный сывороточный α-глобулин, сходный по своим характеристикам с С-реактивным белком (при опухолевых и ревматических заболеваниях, периодической болезни и др.);
- Aβ2М - в составе фибрилл β2-микроглобулин (при хронической почечной недостаточности у больных, находящихся на гемодиализе);
- ATTR – в составе фибрилл транспортный белок транстиретин (при семейных наследственных и старческих формах амилоидоза).
Симптомы амилоидоза
Клинические проявления амилоидоза отличаются многообразием и зависят от выраженности и локализации амилоидных отложений, биохимического состава амилоида, «стажа» заболевания, степени нарушения функции органов. В латентной стадии амилоидоза, когда отложения амилоида могут быть обнаружены только микроскопически, симптоматика отсутствует. По мере развития и прогрессирования функциональной недостаточности того или иного органа нарастают клинические признаки заболевания.
При амилоидозе почек длительно текущая стадия умеренной протеинурии сменяется развитием нефротического синдрома . Переход к развернутой стадии может быть связан с перенесенной интеркуррентной инфекцией, вакцинацией, переохлаждением, обострением основного заболевания. Постепенно нарастают отеки (сначала на ногах, а затем на всем теле), развивается нефрогенная артериальная гипертензия и почечная недостаточность. Возможно возникновение тромбоза почечных вен . Массивная потеря белка сопровождается гипопротеинемией, гиперфибриногенемией, гиперлипидемией, азотемией. В моче обнаруживается микро-, иногда макрогематурия, лейкоцитурия. В целом в течение амилоидоза почек выделяют раннюю безотечную стадию, отечную стадию, уремическую (кахектическую) стадию.
Амилоидоз сердца протекает по типу рестриктивной кардиомиопатии с типичными клиническими признаками – кардиомегалией, аритмией , прогрессирующей сердечной недостаточностью . Больные жалуются на одышку, отеки, слабость, возникающую при незначительных физических нагрузках. Реже при амилоидозе сердца развивается полисерозит (асцит , экссудативный плеврит и перикардит).
Поражение ЖКТ при амилоидозе характеризуется амилоидной инфильтрацией языка (макроглассией), пищевода (ригидностью и нарушением перистальтики), желудка (изжогой, тошнотой), кишечника (запорами, диареей, синдромом мальабсорбции , кишечной непроходимостью). Возможно возникновение желудочно-кишечных кровотечений на различных уровнях. При амилоидной инфильтрации печени развивается гепатомегалия , холестаз , портальная гипертензия . Поражение поджелудочной железы при амилоидозе обычно маскируется под хронический панкреатит .
Амилоидоз кожи протекает с появлением множественных восковидных бляшек (папул, узелков) в области лица, шеи, естественных кожных складок. По внешним признакам поражение кожи может напоминать склеродермию , нейродермит или красный плоский лишай . Для амилоидного поражения опорно-двигательного аппарата типично развитие симметричного полиартрита , запястного туннельного синдрома, плечелопаточного периартрита , миопатии . Отдельные формы амилоидоза, протекающие с вовлечением нервной системы, могут сопровождаться полинейропатией, параличами нижних конечностей, головными болями, головокружением, ортостатической гипотензией, потливостью, деменцией и т. д.
Диагностика
), эндоскопических исследований (ЭГДС , ректороманоскопия). Об амилоидозе следует думать при сочетании протеинурии, лейкоцитурии, цилиндрурии с гипопротеинемией, гиперлипидемией (повышением в крови содержания холестерина, липопротеидов, триглицеридов), гипонатриемией и гипокальциемией, анемией, снижением количества тромбоцитов. Электрофорез сыворотки крови и мочи позволяет определить наличие парапротеинов.Окончательная диагностика амилоидоза возможна после обнаружения амилоидных фибрилл в пораженных тканях. С этой целью может производиться биопсия почки , лимфатических узлов , десен, слизистой оболочки желудка , прямой кишки . Установлению наследственного характера амилоидоза способствует тщательный медико-генетический анализ родословной.
Лечение амилоидоза
Отсутствие полноты знаний об этиологии и патогенезе заболевания обусловливают трудности, связанные с лечением амилоидоза. При вторичном амилоидозе важное значение имеет активная терапия фонового заболевания. Рекомендации по питанию предполагают ограничение приема поваренной соли и белка, включение в рацион сырой печени. Симптоматическая терапия амилоидоза зависит от наличия и выраженности тех или иных клинических проявлений. В качестве патогенетической терапии могут назначаться препараты 4-аминохинолинового ряда (хлорохин), диметилсульфоксид, унитиол, колхицин. Для терапии первичного амилоидоза используются схемы лечения цитостатиками и гормонами (мельфолан+преднизолон, винкристин+доксорубицин+дексаметазон). При развитии ХПН показан гемодиализ или перитонеальный диализ . В отдельных случаях ставится вопрос о трансплантации почек или печени.
Прогноз
Течение амилоидоза носит прогрессирующий, практически необратимый характер. Заболевание может отягощаться амилоидными язвами пищевода и желудка, кровотечениями, печеночной недостаточностью , сахарным диабетом и др. При развитии хронической почечной недостаточности средняя продолжительность жизни больных составляет около 1 года; при развитии сердечной недостаточности – около 4 месяцев. Прогноз вторичного амилоидоза определяется возможностью терапии основного заболевания. Более тяжелое течение амилоидоза отмечается у пожилых пациентов.
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Клинические протоколы МЗ РК - 2016
Амилоидоз (E85)
Нефрология
Общая информация
Краткое описание
Одобрено
Объединенной комиссией по качеству медицинских услуг
Министерства здравоохранения и социального развития Республики Казахстан
от «13» октября 2016 года
Протокол №13
Амилоидоз
- группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида - амилоида.
Соотношение кодов МКБ-10 и МКБ-9
| МКБ-10 | МКБ-9 | ||
| Код | Название | Код | Название |
| E85 | Амилоидоз |
55.23 99.76 |
Закрытая [чрескожная] [пункционная] биопсия почки. Гемодиализ. Терапевтический плазмоферез Экстракорпоральная иммуноадсорбция |
| E85.0 | Наследственный семейный амилоидоз без нейропатии | ||
| E85.1 | Нейропатический наследственный амилоидоз | ||
| E85.2 | Наследственный амилоидоз неуточнённый | ||
| E85.3 | Вторичный системный амилоидоз | ||
| E85.4 | Ограниченный амилоидоз | ||
| E85.8 | Другие формы амилоидоза | ||
| E85.9 | Амилоидоз неуточнённый | ||
Дата разработки/пересмотра протокола: 2016 год.
Пользователи протокола: врачи общей практики, терапевты, гематологи, нефрологи.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С |
Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
Классификация
Типы амилоида и соответствующие формы амилоидоза
:
|
Белок
амилоида |
Белок-предшественник | Клиническая форма амилоидоза |
| АА | SAA-белок | Вторичный амилоидоз при хронических воспалительных заболеваниях, в том числе периодической болезни и синдроме Макла-Уэллса |
| AL | λ, κ-легкие цепи иммуноглобулинов | Амилоидоз при плазмоклеточных дискразиях - идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема |
| ATTR | Транстиретин | Семейные формы полинейропатического, кардиопатического и др. амилоидоза, системный старческий амилоидоз |
| Аβ2М | β2-микроглобулин | Диализный амилоидоз |
| AGel | Гелсолин | Финская семейная амилоидная полинейропатия |
| AApoAI | Аполипопротеин А-I | Амилоидная полинейропатия (III тип, по van Allen, 1956) |
| AFib | Фибриноген | Амилоидная нефропатия |
| Aβ | β-белок | Болезнь Альцгеймера, синдром Дауна, наследственные кровоизлияния в мозг с амилоидозом, Голландия |
| APrP Scr | Прионовый белок | Болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штраусслера-Шейнкера |
| AANF | Предсердный натрийуретический фактор | Изолированный амилоидоз предсердий |
| AIAPP | Амилин | Изолированный амилоидоз в островках Лангерганса при сахарном диабете II типа, Инсулиноме |
| ACal | Прокальцитонин | При медуллярном раке щитовидной железы |
| ACys | Цистатин С | Наследственные кровоизлияния в мозг с амилоидозом, Исландия |
Клиническая классификация амилоидоза
первичный амилоидоз:
· возникающий без явной причины;
· ассоциированный с множественной миеломой;
вторичный амилоидоз
:
· при хронических инфекциях;
· при ревматоидном артрите и других заболеваниях соединительной ткани;
· при онкологических заболеваниях;
семейный (наследственный) амилоидоз:
· при периодической болезни;
· португальский вариант и другие формы семейного амилоидоза;
старческий амилоидоз
локальный амилоидоз
наследственный амилоидоз:
нейропатический
· с поражением нижних конечностей: португальский, японский, шведский и другие типы;
· с поражением верхних конечностей: типы Швейцария-Индиана, Германия-Мэриленд;
нефропатический:
· периодическая болезнь;
· лихорадка и боли в животе у шведов и сицилийцев;
· сочетание сыпи, глухоты и поражения почек;
· поражение почек в сочетании с артериальной гипертензией;
кардиомиопатический:
· датский — прогрессирующая сердечная недостаточность;
· мексиканско-американский — синдром слабости синусового узла, остановка предсердий;
смешанный:
· финский — дистрофия роговицы и поражение черепно-мозговых нервов;
· мозговые инсульты.
Клинические стадии амилоидоза почек
| Стадия | Клиническое проявление |
| 1 | Доклиническая или латентная (бессимптомная) стадия - амилоид присутствует в интермедиарной зоне и по ходу прямых сосудов пирамидок развивается отек и очаги склероза. Стадия длится 3-5 и более лет. В этот период при реактивном амилоидозе преобладают клинические проявления основного заболевания (например, гнойного процесса в легких, туберкулеза, ревматоидного артрита и т. д.). |
| 2 | Протеинурическая (альбуминурическая) стадия - амилоид появляется прежде всего в мезангии, в петлях капилляров, в пирамидах и корковом веществе гломерул, в сосудах. Развиваются склероз и атрофия нефронов, гиперемия и лимфостаз. Почки увеличены и плотны, матово-серо-розового цвета. Протеинурия в начале выражена умеренно, может какой-то период быть даже преходящей, уменьшаться и увеличиваться, но затем становится стойкой (стадия перемежающейся протеинурии). Некоторые исследователи выделяют в этой стадии два периода: селективной и неселективной протеинурии. Продолжительность стадии от 10 до 13 лет. |
| 3 | Нефротическая (отечная, отечно-гипотоническая) стадия - амилоидно-липоидный нефроз - амилоид во всех отделах нефрона. Имеются склероз и амилоидоз мозгового слоя, но корковый слой без выраженных склеротических изменений. Продолжительность стадии до 6 лет. Как в протеинурической, так и в нефротической стадии почки увеличены, плотные (большая сальная почка). Клинически эта стадия проявляется классическим нефротическим синдромом со всеми его признаками: с развитием массивной протеинурии (с потерей белка с мочей более 3-5 граммов в сутки), гипопротеинемии с гипоальбуминемией, гиперхолестеринемии, липидурии с отеками до степени анасарки. В мочевом осадке находят гиалиновые, а по мере нарастания протеинурии - зернистые цилиндры. Возможны микро-и макрогематурия, лейкоцитурия без признаков пиелонефрита. |
| 4 | Уремическая (терминальная, азотемическая) стадия - амилоидная сморщенная почка - уменьшенная в размерах, плотная, с рубцами почка. Хроническая почечная недостаточность мало отличается от таковой при других заболеваниях почек. Считается, что в отличие от гломерулонефрита, при котором наступление ХПН, протекающей с полиурией, может приводить к хотя бы частичному схождению отеков, при амилоидозе азотемия развивается на фоне низкого артериального давления и нефротического синдрома. |
Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОМ УРОВНЕ
Диагностические критерии
Жалобы:
· слабость, повышенная утомляемость;
· головная боль;
· отеки на ногах, руках и лице;
· повышенное артериальное давление;
· тошнота, диарея (понос);
· боли в области сердца;
· боли в мышцах.
Анамнез:
· потеря веса;
· наличие моноклональной гаммапатии неясного генеза;
· хронические воспалительные (гнойные) заболевания;
· хронические инфекции;
· наследственность.
Физикальное обследование
Общий осмотр:
· периорбитальная пурпура (наблюдается в 15% случаев);
· макроглоссия характерно для первичного амилоидоза (AL);
· одышка при физической нагрузке (наблюдается около 40% больных);
· признак наплечника (околосуставная инфильтрация амилоида приводит к ложной гипертрофии и к увеличению объема мускулатуры плечевого пояса и бедра).
Аускультация:
· возможно наличие нарушения сердечного ритма.
Пальпация:
· отеки нижних конечностей, из-за гипоальбуминемии и нефротического синдрома, а также из-за застоя в большом круге кровообращения вследствие рестриктивной кардиомиопатии (наблюдается в 50% случаев);
· увеличение печени и селезенки;
· парестезии (наблюдается около у 15% больных);
· спастические боли в ЖКТ;
· возможно наличие увеличения подчелюстных слюнных желез.
Лабораторные исследования:
· общий анализ крови - анемия, лейкоцитоз, повышение СОЭ;
· общий анализ мочи - протеинурия, микрогематурия, асептическая лейкоцитурия;
· биохимический анализ крови (общий белок, альбумин, Na, Ca, холестерин, сахар в сыворотке крови) - гипопротеинемия (за счёт гипоальбуминемии), гиперглобулинемия, гипонатриемия, гипопротромбинемия, гипокальциемия, гиперхолестеринемия.
· УЗИ органов брюшной полости и почек - визуализируются увеличенные уплотнённые почки (большие жировые почки).

Диагностика (стационар)
ДИАГНОСТИКА НА СТАЦИОНАРНОМ УРОВНЕ
Диагностические критерии на стационарном уровне
Жалобы и анамнез:
см. амбулаторный уровень.
Физикальное обследование: см. амбулаторный уровень.
Лабораторные исследования:
| Диагностический тест | Результат |
|
Сывороточная иммунофиксация
Тест положительный у 60% больных амилоидозом с иммуноглобулином легкой цепи (AL) (6). |
|
|
Иммунофиксация мочи
Тест положительный у 80% больных с AL амилоидозом (6). Обнаружение белка легкой цепи в моче предпологает наличие множественной миеломы и амилоидоза. |
Наличие моноклонального белка |
|
Исследование иммуноглобулинов свободной легкой цепи в сыворотке
Этот относительно новый тест с очень высокой чувствительностью (> 95%) для диагностики AL амилоидоза (10). Имеющийся в продаже антисыворотки к иммуноглобулину легких цепей, AA, и транстиретина, как правило, используются, но возможно, не имеют достаточной специфичности и чувствительности. Во многих случаях, масс - спектроскопия и иммуно-электронная микроскопия необходимы для определения базового типа амилоида. |
Ненормальное соотношение каппа лямбда |
|
Биопсия костного мозга
Биопсия костного мозга проводится у всех пациентов с подозрением на амилоидоз легкой цепи и является отличным источником ткани для диагностики любого пациента с подозрением на амилоидоз. |
Наличие клоновых клеток плазмы |
Инструментальные исследования:
· УЗИ органов брюшной полости и почек - визуализируются увеличенные уплотнённые почки (большие жировые почки).
Диагностический алгоритм амилоидоза почек
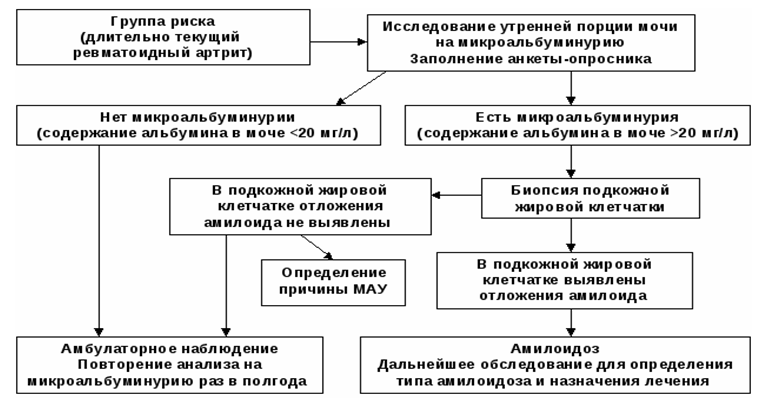
Перечень основных диагностических мероприятий:
· общий анализ крови;
· общий анализ мочи;
· биохимический анализ крови (общий белок, альбумин, Na, Ca, холестерин, сахар в сыворотке крови);
· сывороточная иммунофиксация;
· иммунофиксация мочи;
· исследование иммуноглобулинов свободной легкой цепи в сыворотке;
· биопсия костного мозга.
· УЗИ органов брюшной полости и почек.
Перечень дополнительных диагностических мероприятий
Лабораторные исследования:
| Диагностический тест | Результат |
|
Биопсия тканей:
Для диагностики амилоидоза необходимо, что бы отложения в тканях в биопсийном материале положительно окрашивались по Конго красному (11). Можно увидеть ярко-зеленое двулучепреломление при окрашивании материала Конго красным в поляризованном свете. Биопсийный материал может быть получен из слизистой губ, кожи, десен, подкожной жировой клетчатки, костного мозга, нервов, прямой кишки, почек, печени или сердца. Отложения всегда располагаются внеклеточно и являются аморфными. |
положительное - зеленое двулучепреломление при окрашивании Конго красным |
|
Иммуногистологические исследования амилоидных отложений:
Они позволяют распознавать различные формы системного амилоидоза. |
антисыворотка к иммуноглобулину легкой цепи, AA и транстиретину |
|
Масс - спектроскопия:
Обеспечивает анализ состава амилоидного белка. В настоящее время является золотым стандартом для диагностики типа амилоидоза. |
подтверждает тип белка |
|
Иммуно-электронная микроскопия:
Все формы амилоида под электронным микроскопом волокнистые, жесткие и неветвящиеся. |
амилоиды имеют фибриллярный вид и являются жесткими и неветвящимися. |
|
Генетическое тестирование:
Для исключения наследственного амилоидоза при сомнительных результатах исследования на обнаружение белка моноклональных иммуноглобулинов/свободной легкой цепи амилоида, проведение генетического тестирования обязательно. Гены могут быть исследованы путем прямого секвенирования, и включают в себя следующие гены: TTR, фибриноген, аполипопротеин А1, лизоцим, MEFV (средиземноморская лихорадка) и рецептор 1 фактора некроза опухоли (TNFR1 или TNFRSF1A). MEFV и TNFRSF1A являются наследственными синдромами периодической лихорадки (то есть, потенциальные причины АА амилоидоза), и не являются наследственными амилоидозами сами по себе. |
положительный |
|
Сцинтиграфическое сканирование сывороточного амилоида P (SAP):
В последние годы в клинической практике начали применять метод сцинтиграфии с меченным йодом сывороточным P-компонентом (SAP) для оценки распределения амилоида в организме. |
поглощение на участках отложения амилоида |
|
Общий анализ крови:
Анемия наблюдается в основном у пациентов с почечной недостаточностью или при кровотечениях из ЖКТ. Тромбоцитемия связана с вовлечением в процесс печени и при гиперспленизме. |
как правило, нормальное |
|
Биохимический анализ крови (печеночные и почечные пробы
, показатели метаболического статуса):
Амилоидоз печени характеризуется повышением уровня щелочной фосфатазы. У большинства пациентов на ранней стадии амилоидоза почек сохранен клиренс креатинина, но могут наблюдаться значительные степени гипоальбуминемии из - за потери белка с мочой (нефротический синдром). |
Низкий уровень альбумина; повышение щелочной фосфатазы |
|
Суточная протеинурия (сбор мочи за 24 часа):
Экскреция альбумина > 1 г/сутки у больных с амилоидозом указывает на поражение почек (амилоидоз почек). При уровне протеинурии > 3 г / сутки развивается нефротический синдром. |
повышенный белок в моче |
|
Уровень сывороточного тропонина:
Чувствительный тест для определения повреждения миокарда. Пациенты с выявляемым уровнем тропонина имеют худший прогноз, чем те, у кого он отсутствует (12). |
повышенный |
|
B-тип натрийуретического пептида:
Чувствительное диагностическое исследование на наличие растяжения миокарда и ХСН. Было показано, что имеет важное прогностическое значение в установлении амилоидоза сердца (13). При уровне B-типа натрийуретического пептида > 300 нг / л (> 300 пг / мл) предполагается вовлечение миокарда амилоидом (10). Пациенты с <170 нг / л (<170 пг / мл) имеют значительно более длительную выживаемость, чем пациенты с > 170 нг / л (> 170 пг / мл). |
повышенный |
|
бета-2-микроглобулины:
Является предиктором выживаемости у больных с амилоидозом. При уровне бета-2-микроглобулина > 2,7 мг / л прогноз неблагоприятный (14). |
повышенный |
Инструментальные исследования:
|
ЭКГ:
Должна быть выполнена всем пациентам в рамках оценки вовлечения в процесс сердца. |
нарушение проводимости сердца |
|
Эхокардиограмма (ЭхоКГ):
Клинические признаки сердечной недостаточности у пациентов с амилоидозом сердца наблюдается от 22% до 34% (7). ЭхоКГ обнаруживает высокую частоту отложения амилоида у больных с минимальными симптомами (около 50% случаев AL имеется вовлечение сердца). В последней стадии отмечается снижение фракции выброса. |
диастолическая дисфункция, утолщение межжелудочковой перегородки, снижение фракции выброса |
|
Эхо-допплерография с напряжением:
Показатель степени амилоидной инфильтрации в миокарде. Обладает высокой чувствительностью при обнаружении аномалий, когда нет артериальной гипертензии или клапанной болезни сердца. Растяжение миокарда определяется, как процентное изменение длины волокна миокарда на единицу длины, а скорость зависит от длительности растяжения (15-16). |
Уменьшение продольного сокращения и растяжение миокарда; ограничение наполнения желудочков камер |
|
МРТ сердца:
Магнитно - резонансная релаксометрия улучшает надежность диагностики МРТ и помогает отличить сердечный амилоидоз от гипертрофической кардиомиопатии. |
значительно повышенные времена релаксации T1 и T2 по сравнению с возрастной контрольной группой |
Дифференциальный диагноз
Дифференциальная диагностика амилоидоза почек
| Состояние | Дифференцируемые признаки / симптомы | Дифференцируемые тесты |
| Клинически трудно отличить ГКМ от амилоидоза сердца. |
· ЭхоКГ явяется диагностическим критерием для ГКМ, где выявляется асимметричная гипертрофия межжелудочковой перегородки; · используемая для исключения признаков амилоидоза эхо допплерография с напряжением не указывает на типичные рестриктивные изменения наполнения, выявляемые при амилоидозе; · МРТ позволяет различить 2 синдрома |
|
| Мембранозная гломерулопатия | Клинически одинаковые проявления у пациентов с нефротическим синдромом. | · почечная биопсия не окрашивается Конго красным. |
| Моноклональная гаммапатия неясного генеза (МГНГ) -ассоциированная нейропатия | У пациентов не бывает значительной степени протеинурии, гепатомегалии или кардиомиопатии. | · биопсия икроножного нерва не окрашивается Конго красным. |
| Множественная миелома | Боль в костях, симптомы анемии и почечной недостаточности. |
· обычные рентгеновские снимки показывают литические повреждения кости, компрессионные переломы, диффузный остеопороз; · низкий Hb; · почечная недостаточность. |
| Нефротический синдром | Суточная протеинурия более 3,5 г/сут, отеки, гипоальбуминемия, дислипидемия | · смотрите КП «Нефротический синдром» |
Лечение за рубежом
Пройти лечение в Корее, Израиле, Германии, США
Лечение за рубежом
Получить консультацию по медтуризму
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Альбумин человека (Albumin human) |
| Анакинра (Anakinra) |
| Аторвастатин (Atorvastatin) |
| Бортезомиб (Bortezomib) |
| Валсартан (Valsartan) |
| Гепарин натрия (Heparin sodium) |
| Гидрохлоротиазид (Hydrochlorothiazide) |
| Дексаметазон (Dexamethasone) |
| Дифлунизал (Diflunisal) |
| Индапамид (Indapamide) |
| Инфликсимаб (Infliximab) |
| Канакинумаб (Canakinumab) |
| Кандесартан (Candesartan) |
| Колхицин (Colchicine) |
| Леналидомид (Lenalidomide) |
| Лизиноприл (Lisinopril) |
| Лозартан (Losartan) |
| Мелфалан (Melphalan) |
| Надропарин кальция (Nadroparin calcium) |
| Периндоприл (Perindopril) |
| Рамиприл (Ramipril) |
| Рилонацепт (Rilonacept) |
| Розувастатин (Rosuvastatin) |
| Симвастатин (Simvastatin) |
| Спиронолактон (Spironolactone) |
| Талидомид (Thalidomide) |
| Торасемид (Torasemide) |
| Фозиноприл (Fosinopril) |
| Фуросемид (Furosemide) |
| Циклофосфамид (Cyclophosphamide) |
| Этанерцепт (Etanercept) |
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОМ УРОВНЕ
Тактика лечения: при подозрении диагноза амилоидоз почек, пациента необходимо направить на консультацию нефролога для дальнейшего лечения на стационарном уровне.
Немедикаментозное лечение: нет.
Медикаментозное лечение: нет.
Другие виды лечения: нет.
· консультация нефролога - для постановки диагноза;
· консультация профильных специалистов при наличии сопутствующей патологии.
Профилактические мероприятия:
Первичная профилактика
· первичный амилоидоз почек - профилактических мероприятий нет;
· развитие вторичного амилоидоза из хронического воспалительного состояния непосредственно связано с неконтролируемым воспалением и синтезом сывороточного амилоидного белка печенью. Лечение основного состояния с подавлением воспаления снижает последующий риск вторичного амилоидоза;
· у больных с известными моноклональными гаммапатиями неясного генеза существует риск развития амилоидоза, и рекомендуется мониторинг пациентов для предупреждения развития протеинурии, невропатии, гепатомегалии, или сердечной недостаточности .
Лечение (скорая помощь)
ДИАГНОСТИКА И ЛЕЧЕНИЕ НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ
Диагностические мероприятия:
· оценка состояния с физикальным обследованием (измерение АД, ЧСС, аускультация).
Медикаментозное лечение:
при наличии сопутствующей патологии, смотрите клинический протокол по соответствующим нозологиям.
Лечение (стационар)
ЛЕЧЕНИЕ НА СТАЦИОНАРНОМ УРОВНЕ
Тактика лечения:
Лечение амилоидоза заключается в снижении образования патологического белка и защита органов от его воздействия. При АА-амилоидозе применяются противовоспалительные мероприятия с использованием хирургических методов. При вторичном амилоидозе проводится лечение основного заболевания. При AL амилоидозе проводится подавление клона плазматических клеток, синтезирующий иммуноглобулин легкой цепи. Остановка отложения иммуноглобулинов легкой цепи позволяет организму растворить и вывести излишек амилоида, что предотвращает дальнейшее отложение амилоида. Пациентам с амилоидозом, у которых проводилась биопсия, имеющие висцеральный синдром (то есть, амилоид в сердце, печени, почках, нервах, легком, или кишечнике) рекомендуется трансплантация стволовых клеток/химиотерапии, которое выполняется в специализированном центре для лечения амилоидоза.
Немедикаментозное лечение:
· режим III: постельный при тяжелом состоянии пациента и наличии осложнений, дозированная физическая активность, здоровый образ жизни, отказ от курения и от приема алкоголя;
· Диета: №7. Сбалансированная, адекватное введение белка (1,5-2г/кг), калораж по возрасту, при наличии отеков и АГ - ограничение употребления натрия хлорид (поваренной соли) < 1-2г/сут;
· мониторинг уровня протеинурии по тест-полоскам 1 раз в 1-2 недели, регулярное измерение АД.
· при нарастании протеинурии (рецидиве) определение протеин/креатининового коэффициента (для расчета суточной протеинурии) и коррекция патогенетической терапии;
· при резистентности к проводимой иммуносупрессивной терапии коррекция терапии в условиях стационара.
Медикаментозное лечение:
медикаментозное лечение амилоидоза почек заключается в применении химиотерапии совместно с трансплантацией стволовых клеток.
AL тип амилоидоза.
Впервые диагностированный
AL
амилоидоз
:
· Миелоаблативная химиотерапия с использованием высоких доз мелфалана* и ТСК;
· Миелоаблативная химиотерапия с высокими дозами мелфалана* (после регистрации в РК) и ТСК с использованием идукционной терапии бортезомиба с дексаметазоном;
Показания для ТСК:
· возраст <70 лет;
Противопоказания для ТСК
:
· выраженная сердечная недостаточность;
· общий билирубин >51 мкмоль / л (>3 мг/дл);
· эхо фракция выброса <45%;
· сывороточный тропонин >0,1 мкг/л (>0,1 нг/мл).
NB! Стандартным условием для трансплантации является однократное введение мелфалана*. Это, как правило, дается с учетом риска в дозах от 140 мг/м 2 для пациентов промежуточного риска, до 200 мг/м 2 для пациентов с низким уровнем риска. Сбор стволовых клеток предполагает использование только факторов роста. Минимальный сбор стволовых клеток должно быть 3х10 6 CD34 клеток на кг веса пациента.
NB! Пациенты могут также получить индукционную терапию бортезомибом плюс дексаметазона перед ТСК .
При неполном ответе на ТСК
Химиотерапия после ТСК
Пациентам, которые не достигают нормализацию уровней свободной легкой цепи иммуноглобулина, рекомендуется сочетание мелфалана* и дексаметазона/ циклофосфамида, дексаметазона и талидомида*. Циклы повторяются ежемесячно на срок до 1 года.
Показания для корректирования доза режима ЦДТ:
· возраст >70 лет;
· сердечная недостаточность выше, чем NYHA II;
· со значительной перегрузкой жидкостью организма.
NB! Пациенты после ТСК с частичной чувствительностью начинают полностью отвечать в следствие адъювантной терапии талидомидом* и дексаметазоном . талидомид* довольно токсичен для чувствительных пациентов, и толерантность не образуется у пациентов на предложенную дозу 200 мг/сут, которая обычно используется у пациентов с множественной миеломой. Больные с амилоидозом обычно не переносят дозы талидомида* >50 мг/сут. Талидомид* вызывает значительные неврологические симптомы, запор, кожная сыпь, и сонливость. Терапия обычно не превышает одного года.
Основным комбинациями лекарственным препаратом являются мелфалан* с дексаметазоном/циклофосфамид + дексаметазон + талидомид*.
При неэффективности ТСК, дополнительно рекомендуется проведение химиотерапии:
· мелфалан* и дексаметазон назначается от 6 до 12 месяцев/бортезомиб и дексаметазон назначается с чередованием в одну неделю бортезомиб, а в следующую неделю дексаметазон, в течение до 45 недель/монотерапия бортезомибом.
· при неэффективности предыдущего лечения бортезомибом, можно рассматривать терапию леналидомида с дексаметазоном .
Основным комбинациями лекарственных препаратом являются мелфалан* с Дексаметазоном/Бортезомиб с Дексаметазоном/монотерапия с Бортезомибом Альтернативным комбинациям лекарственных препаратов являются Леналидомид с Дексаметазоном.
AL амилоидоз не подлежащие ТСК.
Впервые диагностированные.
Химиотерапия:
· сочетание мелфалана* с дексаметазоном является основным вариантом для терапии (УД-B) .;
· к дополнительным препаратам относятся циклофосфамид, дексаметазон и талидомид* (УД-C) /леналидомид с дексаметазоном (УД-В) ;
· монотерапию дексаметазоном можно назначать пациентам, которые чувствительны к терапии мелфаланом* (УД-В).
Показания для ТСК:
· возраст <70 лет;
· минимальные признаки сердечной недостаточности (NYHA
· вовлечение амилоидом менее 3-х органов.
NB! На данном этапе рекомендуется определение свободной легкой цепи иммуноглобулина для оценки эффективности лечения и определения продолжительности терапии (от 6 до 12 месяцев).
NB! Основным комбинациями лекарственных препаратов являются мелфалан* с Дексаметазоном.
Альтернативным комбинациям лекарственных препаратов являются циклофосфамид + дексаметазон + талидомид*/леналидомид + дексаметазон/монотерапия с дексаметазоном.
NB! При частичной неэффективности первого курса химиотерапии рекомендуется лечение Бортезомибом.
При возникновении рецидива после основного лечения:
Повторные курсы химиотерапии
рекомендуются месячные курсы мелфалана* и дексаметазона, циклофосфамида в течение от 6 до 12 месяцев, дексаметазона и талидомида* (ЦДT), леналидомида с дексаметазоном ежемесячно на неопределенный срок/следует учитывать курсы с бортезомибом и дексаметазоном.
Основным комбинациями лекарственных препаратов являются мелфалан* с дексаметазоном/циклофосфамид + дексаметазоном + талидомид*/леналидомид с Дексаметазоном/Дексаметазон с Бортезомибом.
Лечение АА типа амилоидоза.
Лечение основного заболевания:
· лечение включает в себя полный контроль основного системного воспалительного процесса;
· при воспалительных артропатиях используются инфликсимаб и этанерцепт со средней продолжительностью лечения 20 месяцев . Блокада интерлейкина-1 возможна при неэффективности/отказа пациента от инфликсимаба или этанерцепта ;
· основные лекарственные препараты инфликсимаб/этанерцепт. Дополнительно могут использоваться анакинра*, цанакинумаб или рилонацепт*.
NB! Если амилоидоз обусловлен локализованной формой болезни Кастельмана, эффективным методом является резекция опухоли.
Семейный вторичный амилоидоз
Семейная средиземноморская лихорадка
рекомендуется применение колхицина* от 0,5 до 0,6 мг 2 раза в день.
Транстиретиновая форма амилоидоза:
Дифлунизал* замедляет прогрессирование нейропатии при множественной мутантной форме транстиретинового амилоидоза . Тафамидис задерживает прогрессирование нейропатии при наследственном Val30Met транстиретиновом амилоидозе . Основные лекарственные препараты дифлунизал* или тафамидис*.
Химиотерапевтические лекарственные средства
Моноклональные антитела
· введение антител под названием 11-1f4 индуцирует облегченный клеточно-опосредованный воспалительный ответ, что приводит к быстрому снижению амилоидом . Этанерцепт рекомендуется для лечения пациентов с поздними стадиями AL (УД-C);
· эпродисат* снижает риск диализ-зависимой почечной недостаточности у пациентов с АА амилоидозом путем дестабилизации каркаса гликозаминогликана амилоидных фибрилл .
Лечение AL типа амилоидоза
| Препараты | Разовая доза | Кратность введения |
| Мелфалан* | 140-200мг/м 2 | Однократно |
| Бортезомид | 1.3 мг/м 2 | 2 раза в неделю по схеме |
| Дексаметазон | 40 мг/сут | 1 раз в день внутрь или в/в по схеме |
| Циклофосфамид | 10 мг/кг | 1 день в/в |
|
Талидомид* |
200 мг/сут |
1 раз в день желательно перед сном и не менее 1 часа после еды |
| Леналидомид | 25 мг/сут | 1 раз в день по схеме |
Лечение АА типа амилоидоза
| Препараты | Разовая доза | Кратность введения |
| Инфликсимаб | 3-10 мг/сут | 1 раз в сутки в/в по схеме |
| Этанерцепт | 50 мг | 1 раз в неделю п/к |
| Анакинра* | 100 мг | 1 раз в день п/к |
| Цанакинумаб | 150-300 мг | 1 раз в 4 недели п/к по схеме |
| Рилонацепт* | 320 мг/сут | По 160 мг п/к на разные участки |
Семейный вторичный амилоидоз
Перечень основных и дополнительных лекарственных средств:
Перечень основных лекарственных средств:
· анакинра*;
· бортезомид;
· дексаметазон;
· дифлунизал*;
· инфликсимаб;
· колхицин*;
· леналидомид;
· мелфалан*;
· рилонацепт*;
· талидомид*;
· цанакинумаб;
· циклофосфамид;
· этанерцепт.
Перечень дополнительных лекарственных средств:
| Нефропротективная терапия - ингибиторы ангиотензин-превращающего фермента | ||
| Препараты | Разовая доза | Кратность введения |
|
Лизиноприл Рамиприл Фозиноприл Периндоприл |
5 - 10 мг 5 - 10 мг 5 - 10 мг 2,5 - 5 мг |
1- 2 раза 1- 2 раза 1- 2 раза 1- 2 раза |
| Нефропротективная терапия - блокаторы ренин-ангиотензина II | ||
|
Лозартан Валсартан Кандесартан |
50-100 мг 80-160 мг 8 - 16 мг |
1-2 раза 1-2 раза 1-2 раза |
| Диуретики | ||
|
Петлевые: фуросемид торасемид Тиазидоподобные: гипотиазид индапамид Антагонисты альдостерона спиронолактон |
1-3мг/кг/сут 5-10 мг 25-100 мг 12,5-25мг/сут |
1 раз 1 раз 1 раз |
| Антикоагулянты | ||
| Гепарин натрия Надропарин кальция Эноксапарин натрия |
2500-5000 МЕ 1000-5000 МЕ 1000-5000 МЕ |
1-2 раза в день 1-2 раза в день 1-2 раза в день |
| Статины | ||
|
Розувастатин Симвастатин Аторвастатин |
10-20 мг 10-20 мг 10-20 мг |
1 раз в день 1 раз в день 1 раз в день |
| Заменители плазмы и других компонентов крови | ||
| Альбумин | 10 % 200 мл, 20% 100 мл | по потребности |
NB! *применение препарата после регистрации в РК
Хирургическое вмешательство:
Трансплантация донорской почки.
Показания:
· развитие ХПН;
· ХГН.
Секвестрэктомия
показания:
· остеомиелит,
Удаление доли лёгкого
показания:
· бронхоэктатический болезнь.
Трансплантация стволовых клеток
Показания для ТСК:
· младше 70 лет;
· с минимальной сердечной недостаточностью (класс
· вовлечение в процесс менее 3-х органов.
NB! Миелоаблативная химиотерапия с использованием мелфалана* с последующей трансплантацией стволовых клеток для восстановления показано для пациентов, с низким риском осложнений, связанных с лечением (УД-A). Преимущества трансплантации для амилоидоза не были достоверно доказаны (УД-B). TСК является единственной терапией, проводимой в данном случае.
Осложнения ТСК:
· внезапная сердечная смерть;
· кровотечение из желудочно - кишечного тракта;
· почечная недостаточность.
Другие виды лечения:
· заместительная почечная терапия (гемодиализ, гемодиафильтрация, перитонеальный диализ).
Показания для консультации специалистов: консультация профильных специалистов при наличии сопутствующей патологии.
Показания для перевода в отделение интенсивной терапии и реанимации:
· неконтролируемое осложнение нефротического синдрома и ОПП;
· внепочечные проявление амилоидоза, требующие госпитализации в отделение интенсивной терапии.
Индикаторы эффективности лечения
· стабилизация/восстановление функции жизненно важных органов;
· предотвращение функциональных нарушений, с увеличением продолжительности жизни больных;
· регресс нефротического синдрома;
· уменьшение протеинурии;
· уменьшение отложений амилоида в тканях.
Дальнейшее ведение:
· амбулаторное наблюдение специалиста по месту жительства;
· УЗИ почек 1 раз в 3 месяца;
· анализы крови, мочи 1 раз 3 месяца.
Госпитализация
Показания для плановой госпитализации:
· верификация диагноза амилоидоз почек;
· наличие нефротического синдрома.
Показания для экстренной госпитализации:
· анасарка (диффузная отечность мягких тканей с преимущественной локализацией в нижней половине туловища);
· олигоанурия (резкое уменьшение количества мочи, выделяемой почками).
Информация
Источники и литература
- Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗСР РК, 2016
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79(4):319-28. 2) Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749-57. 3) BMJ Best Practice: Amyloidosis: BMJ Publishing Group; 2016 . Available from: http://bestpractice.bmj.com/best-practice/monograph/444.html. 4) Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid. 2005;12(1):1-4. 5) Национальные клинические рекомендации по диагностике и лечению АА-и AL-амилоидоза . Научное общество нефрологов России. 2016 . Available from: http://nonr.ru/?page_id=3178. 6) Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45-59. 7) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR. Amyloidosis. Best Pract Res Clin Haematol. 2005;18(4):709-27. 8) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR. Amyloidosis: diagnosis and management. Clin Lymphoma Myeloma. 2005;6(3):208-19. 9) Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-9. 10) Palladini G, Russo P, Bosoni T, Verga L, Sarais G, Lavatelli F, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. 2009;55(3):499-504. 11) Gertz MA. The classification and typing of amyloid deposits. Am J Clin Pathol. 2004;121(6):787-9. 12) Dispenzieri A, Kyle RA, Gertz MA, Therneau TM, Miller WL, Chandrasekaran K, et al. Survival in patients with primary systemic amyloidosis and raised serum cardiac troponins. Lancet. 2003;361(9371):1787-9. 13) Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107(19):2440-5. 14) Zerbini CA, Anderson JJ, Kane KA, Ju ST, Campistol JM, Simms RW, et al. Beta 2 microglobulin serum levels and prediction of survival in AL amyloidosis. Amyloid. 2002;9(4):242-6. 15) Koyama J, Ray-Sequin PA, Falk RH. Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue Doppler echocardiography in patients with AL (primary) cardiac amyloidosis. Circulation. 2003;107(19):2446-52. 16) Weidemann F, Strotmann JM. Use of tissue Doppler imaging to identify and manage systemic diseases. Clin Res Cardiol. 2008;97(2):65-73. 17) Comenzo RL, Gertz MA. Autologous stem cell transplantation for primary systemic amyloidosis. Blood. 2002;99(12):4276-82. 18) Huang X, Wang Q, Chen W, Zeng C, Chen Z, Gong D, et al. Induction therapy with bortezomib and dexamethasone followed by autologous stem cell transplantation versus autologous stem cell transplantation alone in the treatment of renal AL amyloidosis: a randomized controlled trial. BMC Med. 2014;12:2. 19) Cohen AD, Zhou P, Chou J, Teruya-Feldstein J, Reich L, Hassoun H, et al. Risk-adapted autologous stem cell transplantation with adjuvant dexamethasone +/- thalidomide for systemic light-chain amyloidosis: results of a phase II trial. Br J Haematol. 2007;139(2):224-33. 20) Mahmood S, Venner CP, Sachchithanantham S, Lane T, Rannigan L, Foard D, et al. Lenalidomide and dexamethasone for systemic AL amyloidosis following prior treatment with thalidomide or bortezomib regimens. Br J Haematol. 2014;166(6):842-8. 21) Gertz MA, Lacy MQ, Lust JA, Greipp PR, Witzig TE, Kyle RA. Phase II trial of high-dose dexamethasone for untreated patients with primary systemic amyloidosis. Med Oncol. 1999;16(2):104-9. 22) ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636-44. 23) Lidar M, Livneh A. Familial Mediterranean fever: clinical, molecular and management advancements. Neth J Med. 2007;65(9):318-24. 24) Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658-67. 25) Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802-14. 26) Zhang KY, Tung BY, Kowdley KV. Liver transplantation for metabolic liver diseases. Clin Liver Dis. 2007;11(2):265-81. 27) Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar S. Transplantation for amyloidosis. Curr Opin Oncol. 2007;19(2):136-41. 28) Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083-93. 29) Mhaskar R, Kumar A, Behera M, Kharfan-Dabaja MA, Djulbegovic B. Role of high-dose chemotherapy and autologous hematopoietic cell transplantation in primary systemic amyloidosis: a systematic review. Biol Blood Marrow Transplant. 2009;15(8):893-902. 30) Gertz MA. Immunoglobulin light chain amyloidosis: 2014 update on diagnosis, prognosis, and treatment. Am J Hematol. 2014;89(12):1132-40. 31) Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyloidosis: targeted therapy? Haematologica. 2007;92(10):1302-7. 32) Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92(10):1351-8. 33) Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110(2):787-8. 34) Wechalekar AD, Goodman HJ, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood. 2007;109(2):457-64. 35) Dispenzieri A, Lacy MQ, Zeldenrust SR, Hayman SR, Kumar SK, Geyer SM, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109(2):465-70. 36) Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood. 2010;116(23):4777-82. 37) Reece DE, Sanchorawala V, Hegenbart U, Merlini G, Palladini G, Fermand JP, et al. Weekly and twice-weekly bortezomib in patients with systemic AL amyloidosis: results of a phase 1 dose-escalation study. Blood. 2009;114(8):1489-97. 38) Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, Hawkins PN, Perfetti V, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol. 2010;28(6):1031-7. 39) Lamm W, Willenbacher W, Lang A, Zojer N, Muldur E, Ludwig H, et al. Efficacy of the combination of bortezomib and dexamethasone in systemic AL amyloidosis. Ann Hematol. 2011;90(2):201-6. 40) Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356(23):2349-60. 41) Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93-7. 42) Pharmaceuticals M. Study of dexamethasone plus IXAZOMIB (MLN9708) or physicians choice of treatment in relapsed or refractory systemic light chain (AL) amyloidosis (NCT01659658) February 2016 . Available from: https://clinicaltrials.gov/ct2/show/NCT01659658. 43) Solomon A, Weiss DT, Wall JS. Therapeutic potential of chimeric amyloid-reactive monoclonal antibody 11-1F4. Clin Cancer Res. 2003;9(10 Pt 2):3831S-8S. 44) Solomon A, Weiss DT, Wall JS. Immunotherapy in systemic primary (AL) amyloidosis using amyloid-reactive monoclonal antibodies. Cancer Biother Radiopharm. 2003;18(6):853-60. 45) Hussein MA, Juturi JV, Rybicki L, Lutton S, Murphy BR, Karam MA. Etanercept therapy in patients with advanced primary amyloidosis. Med Oncol. 2003;20(3):283-90. 46) Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819-29. 47) Rajkumar SV, Lacy MQ, Kyle RA. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Blood Rev. 2007;21(5):255-65.
Информация
Сокращения, используемые в протоколе
| АВФ | артериовенозная фистула |
| АГ | артериальная гипертензия |
| АД | артериальное давление |
| БКК | блокаторы кальциевых каналов |
| БРА | блокаторы рецепторов ангиотензина |
| БРВ | безрецидивную выживаемость |
| В/в | внутривенное введение |
| ГКМ | Гипертрофическая кардиомиопатия |
| ДБП | диабетическая болезнь почек |
| ДН | диабетическая нефропатия |
| ЖКТ | желудочно-кишечный тракт |
| ЗПТ | заместительная почечная терапия |
| иАПФ | ингибиторы ангиотензинпревращающего фермента |
| КП | клинический протокол |
| МГНГ | Моноклональная гаммапатия неясного генеза |
| МЕ | международная единица |
| МКБ | Международная классификация болезней |
| мРНК | матричная рибонуклеиновая кислота |
| МРТ | - магнитно-резонансная томография |
| НС | нефротический синдром |
| OВ | общую выживаемость |
| ОПП | острое почечное повреждение |
| п/к | подкожно |
| СКФ | скорость клубочковой фильтрации |
| ТСК | трансплантации стволовых клеток |
| УД | уровень достоверности |
| УЗИ | ультразвуковое исследование |
| ХБП | хроническая болезнь почек |
| ХПН | хроническая почечная недостаточность |
| ХСН | хроническая сердечная недостаточность |
| ЧСС | частота сердечной недостаточности |
| ЦДT | циклофосфамид, дексаметазон, талидомид |
| ЭхоКГ | эхокардиограмма |
| Hb | гемоглобин |
| NYHA | New York Heart Association MEFV - Mediterranean fever (средиземноморска лихорадка) |
| SAP | serum amyloid P component (Р компонент сывороточного амилоида) |
| TTR | transteritin (транстеритин) |
| TNFR1 | tumor necrosis factor receptor 1 |
| TNFRSF1A | tumor necrosis factor receptor superfamily, member 1A |
Список разработчиков протокола с указанием квалификационных данных:
1) Туганбекова Салтанат Кенесовна - доктор медицинских наук, профессор АО «Национальный научный медицинский центр», главный терапевт, главный внештатный нефролог МЗСР РК.
2) Гайпов Абдужапар Эркинович - кандидат медицинских наук, ассоциированный профессор, руководитель отдела экстракорпоарльной гемокоррекции АО ННМЦ, врач нефролог первой категории, генеральный секретарь РОО «Общество нефрологов, врачей диализа и трансплантологов».
3) Туребеков Думан Кажибаевич - доктор медицинских наук, доцент, главный нефролог управления здравоохранения г. Астана, врач нефролог первой категории заведующий отделением нефрологии и терапии городской больницы №1 г. Астана.
4) Худайбергенова Махира Сейдуалиевна - главный эксперт по клинической фармакологии АО «Национальный научный центр онкологии и трансплантологии».
Указание на отсутствие конфликта интересов: нет.
Список рецензентов: Айнабекова Баян Алкеновна - доктор медицинских наук, профессор, заведующий кафедрой внутренних болезней по интернатуре и резидентуре, АО «Медицинский университет Астана».
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement, не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement является исключительно информационно-справочным ресурсом. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.
Амилоидоз - заболевание, характеризующееся нарушением обмена веществ, в результате чего образуется новое для организма вещество (амилоид), которое откладывается в органах и нарушает их функции.
Амилоид является сложным гликопротеидом, в котором фибриллярные и глобулярные белки тесно связаны с полисахаридами. Фибрилла амилоида состоит из полипептидных белков; кроме фибриллярного белка, в состав амилоида входит и другой белок - так называемый Р-компонент, одинаковый при всех формах амилоида. Предполагают, что Р-компонент является нормальным сывороточным белком, связанным с амилоидными фибриллами.
Амилоидоз может возникать как осложнение каких-либо заболеваний или развиваться как самостоятельный процесс.
В настоящее время в зависимости от этиологии выделяют несколько форм амилоидоза, имеющих свойственный им биохимический состав амилоидных фибрилл.
Первичный (идиопатический) амилоидоз развивается без видимых причин и поражает различные органы (сердце, почки, кишечник, печень, нервную систему). Биохимическая форма первичного амилоидоза - AL-форма, предшественником такого амилоида являются Ig и легкие цепи иммуноглобулинов. По структуре амилоида и характеру поражения внутренних органов к первичному (идиопатическому) амилои-дозу близок амилоидоз при миеломной болезни, который в настоящее время выделяется в отдельную группу.
Наследственный (генетический) амилоидоз проявляется преимущественным поражением почек, сочетанием поражения почек и нервной системы. В нашей стране наследственный амилоидоз обычно связан с периодической болезнью, которая передается по аутосомно-доми-нантному типу. При этом заболевании амилоидоз может быть единственным проявлением. Биохимическая форма наследственного амилоидоза - AF (предшественником амилоида является преальбумин). В случае периодической болезни биохимическая форма - АА (предшественником является белок SAA).
Приобретенный (вторичный) амилоидоз встречается наиболее часто и развивается при ревматоидном артрите, болезни Бехтерева, туберкулезе, хронических нагноениях - остеомиелите, бронхоэкта-тической болезни, хроническом абсцессе легкого, реже - при неспецифическом язвенном колите, псориазе, лимфогранулематозе, сифилисе, опухолях почки, легкого и др. Биохимическая форма вторичного амилоидоза - АА (его сывороточный предшественник - белок SAA, синтезируемый гепатоцитами).
Старческий амилоидоз - результат инволютивных нарушений обмена белка, обнаруживается в головном мозге, поджелудочной железе, сердце. Биохимическая формула - AS (предшественником является преальбумин).
Локальный амилоидоз развивается без видимых причин, его биохимическая формула - АЕ (предшественник неизвестен).
Патогенез. Хорошо известны лишь отдельные звенья патогенеза (схема 22). Из схемы следует, что под влиянием мутации генов, а также воздействия внешних факторов изменяется иммунитет - уменьшается
количество Т-лимфоцитов. Это приводит к снижению контролирующего их воздействия на В-систему лимфоцитов. В результате уменьшается количество В-клеток, несущих нормальные иммуноглобулины, и увеличивается количество В-клеток, синтезирующих предшественников амилоидной фибриллы. Амилоидобласты в повышенном количестве продуцируют фибриллярный белок, что обусловливает синтез амилоида в большом количестве.
Однако вследствие генетического дефекта амилоидокластов, способствующего снижению их ферментативной активности, достаточной резорбции амилоида не происходит. В результате наблюдается усиленное отложение амилоида в тканях и органах [Мухин Н.А., 1981].
При миеломной болезни амилоидоз развивается в результате повышенной продукции плазмоцитами парапротеина, идущего на построение амилоида. Состав амилоида при разных формах амилоидоза различен, что определяется составом белка амилоидных фибрилл.
При поражении миокарда, периферических нервов (наблюдается преимущественно при идиопатической форме амилоидоза) амилоид откладывается вокруг коллагеновых волокон соединительной ткани. Отложение амилоида вокруг ретикулярных волокон наблюдается при поражении
почек, кишечника, печени, надпочечников, поджелудочной железы (при наследственном и вторичном амилоидозе). Однако возможно сочетание периколлагенового и периретикулярного отложения амилоида, что обеспечивает сочетанные поражения различных органов и систем.
При отложении амилоида в тканях уменьшается количество функционирующих элементов, кардиомиоцитов, гепатоцитов, нервных волокон, почечных клубочков, что в последующем приводит к развитию недостаточности органа.
Так, в сердце амилоид откладывается под эндокардом, в строме и сосудах миокарда, а также по ходу вен в эпикарде. Сердце при этом резко увеличивается в размерах, а количество кардиомиоцитов быстро убывает. Все это приводит к снижению сократительной функции миокарда и сердечной недостаточности, а также нарушениям проводимости и ритма сердца. В головном мозге при старческом амилоидозе амилоид находят в так называемых сенильных бляшках коры, сосудах и оболочках. В коже амилоид откладывается в сосочках и стенках сосудов, что приводит к резкой атрофии эпидермиса. В печени амилоид откладывается между звездчатыми ретикулоэндотелиоцитами синусоидных сосудов, в стенках сосудов, протоков, в соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки атрофируются.
В почках амилоид откладывается в мембране клубочковых капилляров и канальцев нефрона, в мезангии, капиллярных петлях и по ходу ар-териол. По мере накопления амилоида большинство нефронов атрофируется, погибает или замещается соединительной тканью - возникает ами-лоидно-сморщенная почка. Этот процесс можно представить в виде следующей схемы:
протеинурия -> нефротический синдром -> почечная недостаточность.
Соответственно этому в клинической картине выделяют три стадии: 1) начальную (протеинурическую); 2) развернутую (нефротическую); 3) терминальную (азотемическую).
Клиническая картина. Проявления амилоидоза разнообразны и определяются: 1) локализацией амилоида в том или ином органе; 2) степенью выраженности отложений амилоида в органе; 3) основным заболеванием, на фоне которого развился амилоид (при вторичной форме амилоидоза).
При диагностике могут возникнуть затруднения, обусловленные тем, что клинические проявления болезни будут заметны лишь при определенном количестве отложившегося амилоида. В связи с этим неизбежен «латентный» период от момента отложения амилоида до появления симптомов нарушения функционирования органа или системы.
Клиническая картина особенно яркая при поражении почек - наиболее частой локализации отложений амилоида.
На I этапе диагностического поиска в начальной стадии практически никакой информации, свидетельствующей о поражении почек ами-лоидозом, получить не удается. Жалобы больных связаны с основным заболеванием (при вторичном амилоидозе).
В анамнезе имеются сведения о наличии того или иного заболевания (туберкулез легких, остеомиелит, ревматоидный артрит и пр.), его течении, проводившейся терапии. Сами по себе эти сведения не позволяют диагностировать амилоидоз почек, но обращают внимание врача на такую возможность.
В развернутой стадии амилоидоза больные предъявляют жалобы, обусловленные развитием нефротического синдрома, на уменьшение количества мочи, отеки различной распространенности и выраженности, а также жалобы на слабость, отсутствие аппетита, снижение работоспособности. Наряду с ними при вторичном амилоидозе остаются жалобы на проявление основного заболевания.
В терминальной стадии жалобы вызваны развивающейся хронической почечной недостаточностью: снижение аппетита, тошнота, рвота (диспепсические расстройства), головные боли, нарушение сна (нарушения нервной системы), кожный зуд.
На II этапе диагностического поиска в ранней стадии могут обнаруживаться только симптомы, характерные для основного заболевания (при вторичном амилоидозе).
В развернутой стадии выявляют: 1) отеки различной локализации и выраженности; при значительной задержке жидкости в организме могут появляться гидроторакс, гидроперикард, преходящий асцит; 2) артериальную гипертензию (встречается у 12 - 20 % больных амилоидо-зом), дилатацию и гипертрофию левого желудочка; 3) увеличение печени и селезенки вследствие отложения в тканях амилоида (печень и селезенка плотные, безболезненные, с заостренным краем); 4) симптомы основного заболевания (при вторичном амилоидозе).
В терминальной стадии симптоматика определяется выраженностью почечной недостаточности: 1) дистрофический синдром (изменения кожи и слизистых оболочек); 2) серозно-суставной синдром (остеоартропатии, вторичная подагра, сухой перикардит, плеврит); 3) артериальная гипертензия.
На III этапе диагностического поиска при амилоидозе получают наиболее значимую для постановки диагноза информацию, которую можно сгруппировать следующим образом: 1) мочевой синдром; 2) нарушения белкового и липидного обмена; 3) обнаружение отложения амилоидных масс.
Мочевой синдром: 1) протеинурия - важнейший симптом амилоидоза, развивается при всех его формах, но наиболее часто при вторичном амилоидозе. Протеинурия обычно бывает значительной, за сутки выделяется 2 -20 г белка, основную часть которого составляют альбумины. В меньших количествах выделяются глобулины, возможно выведение с мочой сывороточного предшественника амилоида (белок SAA). В терминальной стадии протеинурия сохраняется. В моче можно обнаружить а- и особенно у-гликопротеиды.
Соответственно степени протеинурии обнаруживают гиалиновые и реже зернистые цилиндры. Нечасто диагностируется микрогематурия или лейкоцитурия, однако выраженность ее не соответствует степени протеи-нурии (как это наблюдается при гломерулонефритах). Степени нарушений липидного обмена при амилоидозе соответствует липоидурия с наличием двоякопреломляющих кристаллов в осадке мочи.
Нарушения белкового и липидного обмена: 1) гипопротеинемия в сочетании с гипоальбуминемией и гипер-<Х2- и гипергаммаглобулинемией; 2) гиперхолестеринемия, гипертриглицеридемия, гипербеталипопротеиде-мия. Выраженная диспротеинемия и нарушения липидного обмена приводят к значительному увеличению СОЭ и изменению осадочных проб (тимоловая, сулемовая и др.).
Большое значение для диагноза имеет обнаружение амилоидных масс в органах и тканях: в печени (50 % случаев), селезенке (при пункцион-ной биопсии), слизистой оболочке десны и прямой кишки.
В начальной стадии (протеину рической) при биопсии слизистой оболочки десны чаще получают отрицательный, а прямой кишки - положительный результат; в развернутой стадии (нефротической) в первом случае результаты положительные у половины больных, а во втором - еще чаще.
Наконец, при хронической почечной недостаточности данные биопсии ткани десны положительны более чем в половине наблюдений, а слизистой оболочки прямой кишки почти во всех случаях. Следовательно, биопсию слизистой оболочки десны следует рекомендовать при далеко зашедшем процессе, а прямой кишки - в любой стадии амилоидоза.
При подозрении на идиопатический амилоидоз (протекает чаще с поражением сердца, периферических нервов, реже - почек) целесообразно прежде всего проводить биопсию слизистой оболочки десны, а при вторичном (приобретенном) амилоидозе и наследственных его формах (протекают с преимущественным поражением почек) - биопсию слизистой оболочки прямой кишки.
Ряд других исследований помогает: 1) уточнить диагноз заболевания, на фоне которого развился амилоидоз; 2) оценить функциональное состояние почек (проба Реберга, Зимницкого, уровень креатинина крови).
Течение. Клиническая картина амилоидоза почек имеет особенности, отличающие его от поражения почек иного происхождения: 1) неф-ротический синдром развивается постепенно и нередко после длительной стадии протеинурии, отличается упорным течением, отеки часто резистентны к различным мочегонным средствам. При ХГН нефротический синдром возникает, как правило, уже в начале болезни и в дальнейшем часто рецидивирует; 2) артериальная гипертензия наблюдается нечасто, даже в стадии хронической почечной недостаточности; 3) при первичном амилоидозе хроническая почечная недостаточность протекает более доброкачественно в отличие от вторичного амилоидоза или ХГН (вследствие меньшей тяжести поражения клубочков по сравнению со вторичными формами амилоидоза); 4) течение вторичного амилоидоза в значительной степени зависит от основного заболевания, при частых обострениях которого возможно значительное прогрессирование амилоидоза.
Осложнения. При амилоидозе в 2 -5 % случаев развиваются:
1) тромбоз почечных вен (при вторичном амилоидозе), что проявля
ется гематурией и болями в поясничной области, нарастанием протеину
рии и падением диуреза;
2) интеркуррентная инфекция;
3) фибринозно-гнойный перитонит, появление которого сопровожда
ется резким увеличением асцита.
Диагностика. Клинические проявления амилоидоза неспецифичны. Каждый из симптомов (отеки, протеинурия, артериальная гипертензия) могут встречаться при различных заболеваниях почек. Единственным методом достоверной диагностики амилоидоза является биопсия органа (почка, печень, слизистая оболочка прямой кишки или десны), однако она не всегда выполнима. Поэтому в большинстве случаев приходится ориентироваться на клинические проявления патологического процесса.
Наличие заболевания, при котором может развиться вторичный
амилоидоз (клинические или анамнестические признаки).
Появление и прогрессирование протеинурии или возникновение
нефротического синдрома.
Заболевание, при котором может развиться амилоидоз, отсутствует,
однако имеется протеинурия или нефротический синдром.
Наличие стойкой тяжелой сердечной недостаточности, синдрома не
достаточности всасывания, полинейропатии (если при этом послед
ние три синдрома трудно объяснить другими причинами).
Можно предположить наличие амилоидоза при следующих лабораторных признаках нефротического синдрома (который, как известно, может развиваться и при других заболеваниях почек):
а) выраженная диспротеинемия + гипоальбуминемия + гипер-ссг- и ги-
пергаммаглобул инемия;
б) повышение уровня аг-гликопротеида, р-липопротеидов;
в) появление в моче а- и особенно у-гликопротеидов и а-липопро-
теидов.
Во всех случаях вероятность развития амилоидоза увеличивается при обнаружении гепато- и спленомегалии, а также изменений сердца, свойственных амилоидозу (в подобных случаях речь идет об идиопатическом генерализованном амилоидозе).
Следовательно, диагноз амилоидоза почек с достаточной уверенностью можно поставить в развернутой (нефротической) или терминальной стадии, тогда как в начальной (протеинурической) стадии это сделать много труднее. В этих случаях преходящую или постоянную протеинурию приходится дифференцировать от гломерулонефритов (острого, хронического). При этом следует учитывать:
1) более медленное прогрессирование поражения почек при амилои
дозе;
2) отсутствие при амилоидозе четкой связи с простудными заболева
ниями;
3) постоянное наличие при гломерулонефритах микрогематурии (при
амилоидозе в 20 % случаев).
Иногда правильный диагноз можно поставить лишь после периода длительного наблюдения за больным. Вопрос решается значительно быстрее, если имеется возможность произвести пункционную биопсию почки.
Формулировка развернутого клинического диагноза амилоидоза учитывает следующие компоненты: 1) форму амилоидоза; 2) стадию амилоидоза (протеинурическая, нефротическая, терминальная); 3) функциональное состояние почек (отсутствие или наличие почечной недостаточности, степень ее выраженности); 4) основное заболевание (при вторичном амилоидозе); 5) состояние других органов (сердце, печень, нервная система и пр.) при идиопатическом (первичном) амилоидозе.
Лечение. Все еще остается нерешенной проблема лечения амилоидоза, так как не выяснены причины, приводящие к усилению амилоидогене-за и недостаточности его резорбции. Тем не менее возможно проведение серии лечебных мероприятий, улучшающих состояние больного. В настоящее время лечение больного амилоидозом проводится с учетом: 1) воздействия на основное заболевание, на фоне которого развился амилоидоз
(вторичный); 2) воздействия на механизмы патогенеза; 3) воздействия на основные клинические синдромы.
Воздействие на основное заболевание, на фоне которого развивает
ся вторичный амилоидоз, необходимо ввиду того, что частые обо
стрения или высокая активность патологического процесса ведут к
прогрессированию амилоидоза.
Это воздействие заключается в следующем:
а) при хронических инфекциях (туберкулез, сифилис) необходима
длительная специфическая терапия;
б) при хронических неспецифических заболеваниях легких - ком
плексная терапия с применением антибиотиков, бронхиального дренажа, а
при необходимости и оперативное вмешательство (например, при хрони
ческом абсцессе легкого);
в) при системных заболеваниях соединительной ткани, например при
ревматоидном артрите, показана комплексная терапия, включающая на
значение базисных препаратов (D-пеницилламин, соли золота, аминохи-
нолиновые препараты).
Воздействие на механизмы патогенеза предполагает уменьшение
синтеза амилоида:
а) ежедневный прием 80-120 г сырой печени в течение 6-12 мес
приводит к снижению протеинурии, уменьшению размеров пече
ни и селезенки;
б) аминохинолиновые препараты (хингамин, или делагил, по
0,25 - 0,5 г в день в течение многих месяцев и даже лет) снижа
ют прогрессирование процесса. По-видимому, эти средства влия
ют на синтез амилоидных фибрилл. Лечение эффективно только
в ранних стадиях амилоидоза; при далеко зашедшем процессе
(развернутый нефротический синдром, почечная недостаточ
ность) назначение этих препаратов нецелесообразно;
в) при развитии амилоидоза вследствие периодической болезни ре
комендуется колхицин;
г) при первичном амилоидозе назначают также мелфалан, угнетаю
щий функцию некоторых клонов лимфоцитов, в частности син
тезирующих легкие цепи иммуноглобулинов, участвующих в
формировании амилоидной фибриллы (это имеет отношение и к
амилоидозу, развивающемуся при миеломной болезни).
Воздействие на основные клинические синдромы предусматривает
ликвидацию отеков, АГ, а также мероприятия, направленные на
борьбу с развивающейся почечной недостаточностью:
а) при развитии нефротического синдрома и выраженных отеков
необходимо достаточное содержание в пище белка, снижение по
варенной соли, а также введение цельной крови или эритроцит-
ной массы (особенно при наличии анемии), осторожное приме
нение мочегонных средств;
б) артериальная гипертензия нечасто встречается при амилоидозе,
однако, когда она достигает высоких цифр, необходимо назначе
ние гипотензивных средств различного типа;
в) при развитии почечной недостаточности лечение проводится по общепринятому плану (ограничение белка в пище, достаточное введение жидкости, коррекция минерального обмена). При почечной недостаточности, обусловленной амилоидозом, возможно применение гемодиализа и трансплантации почек.
Прогноз. Длительность протеинурического периода установить трудно, однако после его выявления обычно через 3 года развиваются отеки, на фоне которых быстро возникает ХПН. Все это делает прогноз достаточно серьезным.
Профилактика. При идиопатическом и генетическом амилоидозе меры первичной профилактики неизвестны. При вторичном амилоидозе профилактика состоит в лечении заболеваний, ведущих к развитию амилоид оза.



