متلازمة أولبرايت. متلازمة أولبرايت: الأسباب والأعراض والعلاج. يحدث هذا المرض في الغالب عند الفتيات.
متلازمة ماكيون أولبرايت هو مرض غير متجانس تم وصفه بشكل مستقل من قبل طبيب الأطفال الأمريكي د.ج. المرض نادر خلقي ولكنه ليس وراثيًا ، مصحوبًا باضطرابات التمثيل الغذائي وعلامات التطور الجنسي المبكر وتشوهات الهيكل العظمي. لوحظت المظاهر السريرية الأولى في مرحلة الطفولة المبكرة أو المراهقة في شكل اضطرابات في النمو ، خلل التنسج الليفي متعدد التكوينات ، تصبغ الجلد مثل "القهوة مع الحليب" وتشوهات الغدد الصماء ، بما في ذلك ، متلازمة كوشينغ , .
من المرجح أن يتم تشخيص متلازمة أولبرايت ماكيون عند الفتيات بنسبة 10 مرات أكثر من الأولاد. تلاحظ ويكيبيديا أن معدل الحدوث في البشر هو 1 لكل 100 ألف ، أو لكل مليون فرد.
طريقة تطور المرض
نتيجة لطفرة ما بعد اللواقح للجين GNAS1تشارك في إشارات بروتين الخلية GS-α، توفير اقتران مستقبلات الهرمونات اللوتينية وهرمونات تحفيز الجريب مع الجزيئات أدينيلات سيكليس في الهياكل الخلوية للمبايض. يعمل التأثير المستمر للبروتين الطافر على تنشيط محلقة الأدينيلات ويزيد داخل الخلايا معسكر ، ويحفز أيضًا زيادة إفراز الهرمونات الجنسية الأنثوية الستيرويدية عن طريق الأكياس الجريبية المنتجة للإستروجين ، والتي تسبب التطور الجنسي المبكر حتى في حالة عدم وجودها. لوحظ نفس النمط ويلاحظ "الإنتاج المستقل" مع هرمونات الغدة الدرقية ، و. تطور علم الأمراض ناتج عن تكوين الخلايا التي تحتوي بالفعل على بروتينات متحولة.
بالإضافة إلى ذلك ، تؤدي الزيادة في مستوى cAMP إلى زيادة الإنتاج وتشكيل البقع lentious ، وفرط تصبغ بؤري على خلفية القيم الطبيعية و الميلانوتروبين .
خلل التنسج الكيسي الليفي نتيجة للتكلس المعزز تحت تأثير هرمون الاستروجين ، يتم إجراؤه عن طريق استبدال الهياكل العظمية الطبيعية والتكلسات المتكونة بكتل ليفية ، في حين يتم إزعاج توازن عمليات ارتشاف العظام وتجديدها ، وتكاثر الخلايا اللحمية اللحمية. لوحظ تشكيل بؤر الدمار والأورام الكاذبة.
تصنيف
يختلف مرض أولبرايت ، اعتمادًا على شدة الدورة ، يميزون:
- مرض كامن - بدون أي علامات لأمراض العظام أو الغدد الصماء ، وكذلك بدون أعراض تصبغ غير عادي ؛
- أشكال غير مكتملة من المتلازمة ، على سبيل المثال ، في عملية التسبب في الأنسجة العظمية ، من الممكن حدوث تغييرات في الغدد الصماء و قصور جارات الدرقية الكاذب ولكن لا توجد مظاهر جلدية
- قصور جارات الدرقية الكاذب أو وراثي حثل عظم أولبرايت - مرض يؤثر أيضًا على أنسجة العظام ، لكن الآلية والمسببات مختلفة - ينتج علم الأمراض عن انتهاك لمقاومة هرمون الغدة الدرقية ، استقلاب الكالسيوم والفوسفور. يتجلى قصور الدريقات الكاذب أيضًا في مرحلة الطفولة ويتم التعبير عنه في شكل قصر القامة ، والوجه المستدير ، والتخلف العقلي ، قصور الغدد التناسلية ، إلخ.

الأسباب
تحدث متلازمة أولبرايت ماكيون نتيجة الاضطرابات الوراثية في المراحل المبكرة من التطور الجنيني. حتى الآن ، لم يتم تحديد نوع الميراث ، وكانت جميع الحالات الموصوفة متفرقة.
أعراض
تتجلى الصورة السريرية للمرض في شكل مجمع سمبي كامل ، يتكون من:
- التطور الجنسي المبكر المستقل عن الغدد التناسلية ، يتطور في وقت متأخر وببطء أكثر من الأشكال الأخرى من البلوغ المبكر ؛
- تصبغ غير مكتمل للجلد - النمشة ، عادة ما تظهر البقع ذات الحدود الواضحة للون البني الفاتح منذ الولادة على الصدر والظهر وأسفل الظهر والوركين وفي أماكن تشوهات العظام ؛
- اضطرابات الغدد الصماء التي تسبب فقدان الوزن غير المبرر ، وأعراض العين فرط نشاط الغدة الدرقية ;
- خلل التنسج الليفي متعدد التوضعات , وغالبًا ما تنطوي على عظام طويلة في هذه العملية ، تظهر الاضطرابات نفسها في شكل ملامح وجه مشوهة ، وبنية بدنية أكثر استطالة ، ومعصمين ملتويين وغير متماثلين ، وقصر المعصمين ، مما يجعل من الصعب الحركة أثناء المشي ، ويسبب العرج ، والألم الشديد وزيادة احتمال حدوث كسور مرضية.

تبدأ العلامات الأولى للبلوغ المبكر مع نزيف الرحم الذي ليس له طبيعة دورية وينتج عن زيادات قصيرة المدى في تركيزات هرمون الاستروجين وهرمونات موجهة الغدد التناسلية في مجرى الدم. تثير هذه التقلبات تكوين أكياس في المبايض. في الأطفال الذين تتراوح أعمارهم بين 5-10 سنوات ، نمو الثدي المبكر و التثدي . لا تؤدي مسبباته اللطيفة إلى تكوين خصائص جنسية ثانوية - متلازمة ماكيون أولبرايت برايتسيفلا يسبب نمو الشعر الجنسي المبكر أو تمايز الشكل. البلوغ الحقيقي ممكن بعد نهاية سن البلوغ.
التحليلات والتشخيصات
إذا اشتبه في متلازمة ماكيون ، يتم إجراء أولبرايت:
- دراسات الأشعة السينية التي تظهر الأكياس الكاذبة ، الآفات القطاعية في أي جزء من الهيكل العظمي - في كثير من الأحيان في الأطراف السفلية ، وفي حالات نادرة - في حزام الكتف العلوي والجمجمة ؛
- تسمح لك الموجات فوق الصوتية لتجويف البطن بالكشف ؛
- الاختبارات المعملية للكشف عن المستويات المرتفعة من الفوسفاتيز القلوي في مجرى الدم ، وانخفاض مستويات الجونادوتروبين في اختبارات البول ، وزيادة الإخراج الهرمون الملوتن , 17-هيدروكسي و 17-هيدروكسي الستيرويدات القشرية .
علاج او معاملة
العلاج عرضي ، وله صعوباته الخاصة ، اعتمادًا على الخصائص الفردية والصورة السريرية. نظائرها الغدد التناسلية ليس لها تأثير علاجي ، لأن عملية إفراز هرمون الاستروجين لا تسببها زيادة في الجونادوتروبين. لوقف عملية تكوين الستيرويد الغدد التناسلية عند الذكور ، يمكن أن تكون فعالة أيضًا. أثناء العلاج ميدروكسي بروجستيرون أسيتات التطوير الممكن نقص كالسيوم الدم لذلك ، إذا كانت عملية تطور البلوغ المبكر مصحوبة بآفات متعددة وخيمة في أنسجة العظام ، فيجب أن يوصف العلاج بحذر.
من أجل القهر فرط الاستروجين ويمكن وصف أدوية إطالة الهيكل العظمي. لمواجهة خلل التنسج العظمي ، يجب أن تدخل:
- البايفوسفونيت ;
- مثبطات مناهضة ناقضات العظم.
يحتاج المرضى بانتظام كل 3 أشهر. قم بزيارة أخصائي الغدد الصماء لتصحيح مشاكل الغدة الدرقية والغدد الأخرى.
الأطباء
الأدوية
- - على الرغم من كونه معروفًا باسم عقار مضاد للفطريات ، إلا أنه في الجرعات العالية يكون فعالًا في مظاهر تشبه كوشينو. يمكن أن تصل الجرعة اليومية ، مقسمة إلى 2-3 جرعات ، إلى 1200 مجم.
- ميدروكسي بروجستيرون - له تأثيرات أندروجينية ومضادة للأورام وابتنائية. هناك طرق مختلفة لإدخالها في الجسم بجرعات يحددها الطبيب المعالج بشكل فردي.
- حمض الباميدرونيك - الدواء قادر على منع تدمير العظام. يمكن إعطاؤه عن طريق بلعة في الوريد ولكن لا ينبغي دمجه مع العلاج بالبايفوسونات الأخرى.
- - البيسفوسفونات ، الذي يساهم في تكوين العظام ذات التركيب النسيجي الطبيعي وترميمها وزيادة كثافة المعادن. يجب تناول الدواء عن طريق الفم من الداخل - قرص واحد في الأسبوع.
الإجراءات والعمليات
- استئصال المبيض - قد يوصى بالتعقيم في حالات التطور المفرط لنمو فرط البلوغ.
- إعادة البناء الجراحي لتشوه عظام الهيكل العظمي والجمجمة.
تنبؤ بالمناخ
في السنوات الأخيرة ، بدأ الحديث عن متلازمة ماكيون أولبرايت ومشكلات علاجها أكثر فأكثر ، وبدأ الأشخاص الذين يعانون من هذا المرض يعلنون أنفسهم ويكشفون ملامح الحياة اليومية والاجتماعية في المدونات الشخصية ومقاطع الفيديو. بفضل التقنيات الحديثة ، على الرغم من عدم استدامة علم الأمراض ، زادت جودة حياة هؤلاء الأشخاص بشكل كبير ، والآن يمكنهم إيجاد طريقة للتعبير عن أنفسهم وحتى التمثيل في الأفلام ، على سبيل المثال ، مثل الممثل الأوروغواياني موريسيو سارافيا.
قائمة المصادر
- ماكيون. D. J. Osteitis fibrosa cystica: حالة فتاة تبلغ من العمر تسع سنوات تظهر أيضًا البلوغ المبكر ، وتصبغ الجلد المتعدد وفرط نشاط الغدة الدرقية. المجلة الأمريكية لأمراض الأطفال ، شيكاغو ، 1936 ؛ 52: 743-747.
- Mandel B. R. أساسيات علم الوراثة الحديث. الدورة التعليمية. م: فلينتا ، 2017. - 305 ص.
- كولاكوف في ، مانوخينا آي بي ، سافيليفا ج. أمراض النساء: نات. اليدين م: جيوتار ميديا ، 2011. - 461 ص.
هذه المعلومات مخصصة لمتخصصي الرعاية الصحية والأدوية. يجب على المرضى عدم استخدام هذه المعلومات كنصائح أو توصيات طبية.
حالة متلازمة ماكيون أولبرايت برايتسيف
أخصائي جراحة العظام والرضوض من أعلى فئة Bagrinovskaya Irina Leibovna
مستوصف مدينة الأطفال بموسكو رقم 110
الكلمات المفتاحية: فرط التصبغ / البنات / الأطفال / النمشة / الطفرة / الكسر المرضي / خلل التنسج العظمي المتعدد / البلوغ المبكر / متلازمة ماكيون أولبرايت برايتسيف / كيس جرابي.
متلازمة ماكيون-أولبرايت-بريتسيف (متلازمة ماكيون-أولبرايت-برايتسيف) أو متلازمة أولبرايت-ماكيون-ستيرنبرغ ، كما هو مدرج تحت الرمز Q78.1 في أمراض التصنيف الدولي العاشر. يرجع أول مؤشر على هذا المرض إلى الجراح الروسي والسوفيتي ف.ر. Braitsev ، الذي في عام 1907. وصف مرض الكيس الليفي في الفك ( خلل التنسج الليفي كثرة الكيسات) في الأطفال الذين يعانون من بقع الشيخوخة. ومع ذلك ، لم يرد اسم المتلازمة إلا بعد منشورات طبيب الأطفال الأمريكي دي جي ماكيون في عام 1936. وطبيب الغدد الصماء الأمريكي ف.ألبرايت بالتعاون مع عالم الأمراض دبليو ستيرنبرغ في عام 1937.
المظاهر السريرية للمتلازمة:
- بقع غير متماثلة مفرطة التصبغ من لون القهوة مع الحليب (lentigo) ، عادة على جلد الصدر والظهر ومنطقة أسفل الظهر والفخذين ؛
- اعتلالات الغدد الصماء ، وغالبًا ما تكون النمو الجنسي المبكر ؛
- خلل التنسج العظمي الكيسي الليفي متعدد التوضعات.
تحدث متلازمة MRD بسبب طفرة جسدية في جين GNASI ، الموجود على الذراع الطويلة للكروموسوم العشرين ، في المراحل المبكرة من التطور الجنيني ، وبالتالي فهو غير موروث. نتيجة لذلك ، يتم تشكيل مستنسخات من الخلايا الطافرة. يقوم جين GNASI على وجه التحديد بترميز الوحدة الفرعية α لبروتين G (Gαs) ، الذي يتوسط في تحويل الأدينوزين أحادي الفوسفات الدوري (cAMP) ، الذي ينظم النظام الهرموني البشري.
مع زيادة مستوى cAMP ، يزداد إنتاج الميلانين مع القيم الطبيعية لـ ACTH وهرمون تحفيز الميلانين ، مما يؤدي إلى تكوين بقع عدسية.
نظرًا لأن Gαs تعمل كوسيط في نقل الإشارات من LH و FSH ، فإن الخلايا الطافرة في الغدد التناسلية تُظهر تفاعلًا لا إراديًا متزايدًا بشكل حاد ، مما يؤدي إلى ظهور الأكياس المنتجة للإستروجين المسامي عند الفتيات والتطور الجنسي المبكر. يتم توزيع الخلايا المتحولة بشكل غير متساو في الغدد التناسلية. في الفتيات ، يمكن توطينهن في مبيض واحد أو مبيضين ، مما يتسبب في التكوين الدوري لكيسات نشطة أحادية الجانب أو ثنائية مع انحدارها اللاحق.
تبدأ Gαs النشطة في اختلال التوازن بين ارتشاف العظام وتجديدها ، وتكاثر الخلايا اللحمية غير الناضجة ، مما يؤدي إلى استبدال بنية العظام الطبيعية بكتل ليفية وتشكيل بؤر تدمير في شكل خراجات.
من خلال التوسط في تأثيرات هرمون الغدة الجار درقية (PTH) ، تشارك Gαs في الحفاظ على توازن العظام واستقلاب الكالسيوم والفوسفور. PTH هو العامل الرئيسي المسؤول عن استتباب الكالسيوم. يُغسل الكالسيوم من العظام ويترسب على شكل تكلسات في الدهون تحت الجلد والعضلات والأعضاء الداخلية وبطانة الأوعية الدموية. بمرور الوقت ، يتم استبدال الأنسجة العظمية بنسيج ليفي. تشوهات الهيكل العظمي تعيق حركة المرضى وتسبب ألما شديدا. يؤدي انخفاض فيتامين د في الدم إلى حدوث خلل في امتصاص الكالسيوم في الأمعاء. يحفز نقص كالسيوم الدم على إطلاق كمية أكبر من هرمون الغدة الدرقية في الدم وزيادة ارتشاح الكالسيوم من العظام ، مما يؤدي إلى الإصابة بتلين العظام الشديد.
في عملية نمو الجسم وزيادة حمل الوزن ، ينحني الجزء المرضي من العظم. في عدد من المرضى ، يكون عظم الفخذ مشوهًا ، ونتيجة للكسور المرضية ، يتخذ شكل انحناء الراعي. في بعض المرضى ، يؤدي تشوه العظام الشديد إلى الإعاقة.
في وجود الخلايا الطافرة في الغدد الصماء الأخرى ، قد يحدث تضخم الغدة الدرقية متعدد العقيدات ، والكساح الناقص الفوسفات ، ومتلازمة كوشينغ ، ضخامة النهايات ، وما إلى ذلك. مع هزيمة عضلات القلب ، لوحظ عدم انتظام دقات القلب. الآفات المحتملة للجهاز الهضمي ، مثل التهاب الكبد الصفراوي ، الاورام الحميدة المعدية المعوية ، الارتجاع المعدي المعوي.
يتأثر كلا الجنسين بهذا المرض ، لكنه يحدث في كثير من الأحيان عند الفتيات أكثر من الفتيان. البلوغ المبكر عند الفتيات أكثر شيوعًا 9 مرات من الفتيان.
متلازمة MRD هي مرض نادر (يتيم). يختلف تواتر الحدوث في العالم من 1: 100،000 إلى 1: 1،000،000 في عموم السكان.
الحالية والمتوقعة. المرض غير متجانس سريريا. جنبا إلى جنب مع حالات ثالوث العلامات الكلاسيكي ، هناك أشكال غير نمطية وغير كاملة من المتلازمة. كما أن شدة الأعراض وشدة المرض متغيرة بدرجة كبيرة. مع تقدم العمر ، تتطور أمراض أنسجة العظام ، ومع ذلك ، مع الأشكال الممحاة ، يتباطأ التقدم مع بداية سن البلوغ. إن تطور أمراض العظام فقط في التردد يتجاوز مجمع الأعراض الكامل بمقدار 30-40 مرة.
المراقبة السريرية. تم إدخال فتاة تبلغ من العمر 12 عامًا في عيادة جراحة العظام والصدمات في عيادة مدينة الأطفال في موسكو رقم 110 في 31 مايو 2017.
الشكاوى في القبول لألم في الساقين ، تتفاقم بعد المشي لمسافات طويلة أو الرقص. وبحسب المريضة ، فإن الألم شديد لدرجة أنه من المستحيل تحمله ، وعليها التوقف والجلوس في أي مكان. يعتبر نفسه مريضا لمدة 3 أشهر.
تاريخ العائلة: الوالدان روسيان الجنسية ، لا تربطهما صلة قرابة. لم تكن هناك حالات لأمراض عقلية وعصبية وتأخر في النمو في النسب. الأشقاء ، أخت تبلغ من العمر 15 عامًا ، تتمتع بصحة جيدة وفقًا لوالدتها.
سوابق الحياة: طفل من الحمل الثاني ، ولادة عند الأوان ، فسيولوجي ، وزن عند الولادة 3500 جم ، ارتفاع 51 سم ، صرخ على الفور ، أحرز أبغار 8/9 نقاط. بدأت تمسك برأسها في عمر شهرين ، وتجلس في عمر 7 أشهر ، وتمشي بشكل مستقل في عمر عام ، وترضع حتى 5 أشهر. تم تلقيحها حسب العمر. الأمراض المعدية: السارس ، جدري الماء.
بيانات من دراسة موضوعية. الارتفاع 158 سم ، الوزن 40 كجم. ضغط الدم 120/75 مم زئبق ، معدل ضربات القلب 78 / دقيقة. الحالة العامة مرضية. الحالة الوظيفية: يمشي ، مشية لا تنزعج. اللياقة البدنية صحيحة ونمطية. على الكتفين الأيمن والأيسر ، على السطح الجانبي من الجسم على اليمين ، توجد بقع متضخمة صغيرة واحدة بنية اللون ذات حواف غير متساوية يصل قطرها إلى 20 مم. يتم تطوير الدهون تحت الجلد بشكل معتدل ، وتوزيعها بالتساوي. الأغشية المخاطية المرئية نظيفة وردية ورطبة. العقد الليمفاوية المحيطية مفردة ، صغيرة ، متحركة ، غير مؤلمة. تم تطوير كتلة العضلات بشكل كافٍ. هناك عدم تناسق طفيف في مثلثات الخصر ، وعدم تناسق الحوض إلى اليسار. ينحني محور العمود الفقري في المستوى الأمامي الأيسر في منطقة أسفل الظهر. محور الأطراف السفلية صحيح. الحركات في المفاصل كاملة وغير مؤلمة ويلاحظ فرط حركة الرضفة. قدم مسطحة مستعرضة ، تشوه أروح في أصابع القدم الأولى.
التشخيص الأولي: "العظام من أصل غير واضح. الجنف من الدرجة الأولى. قدم مسطحة مستعرضة. تشوه أروح أصابع القدم الأولى.
اختبار الدم السريري: الهيموغلوبين 128 جم / لتر ، الكريات البيض 5.2 × 10 9 / لتر ، طعنة 1٪ ، مجزأة 54٪ ، الحمضات 1٪ ، الخلايا الليمفاوية 36٪ ، وحيدات 8٪ ، الصفائح الدموية 218 × l0 9 / لتر ، مؤشر اللون 0 ، 9 ٪ ، معدل ترسيب كرات الدم الحمراء 8 مم / ساعة.
ECG بدون ميزات.
التصوير الشعاعي للساق ومفاصل الركبة 31/05/2017. تم الكشف عن خلل التنسج الليفي في قصبة الساق. وفقًا لنتائج الفحص بالأشعة المقطعية للأطراف السفلية بتاريخ 1 يونيو 2017 ، تم الكشف عن شكل متعدد التكوينات من خلل التنسج الليفي لعظام الأطراف السفلية.

تقرر إجراء التشخيص التفريقي بين الأشكال المختلفة لخلل التنسج الليفي متعدد التكوينات. في الموعد الثاني ، تم الكشف عن تفاصيل سوابق الحياة ، التي كانت الأم قد أخفتها من قبل. لم يتم تضمين المقتطفات في بطاقة العيادة الخارجية واحتفظت بها الأم في المنزل.
استمرار سوابق الحياة والمرض: في سن ستة أشهر ، لوحظ وجود الثيلارش المعزول. في الثانية من العمر ، ثعلبة ثنائية ، كتل الثدي على اليمين بقطر 3.5 سم ، على اليسار 3 سم. الصيغة الجنسية هي Ma2P0Ax0. لمدة 12 شهرًا ، تكررت الزيادة في الغدد الثديية 5 مرات ، وكانت مدة التقدم 3 أسابيع وكان الانحدار أسبوعين. تمت استشارته وفحصه من قبل أخصائي الغدد الصماء في معهد طب الغدد الصماء لدى الأطفال ، مركز أبحاث الغدد الصماء التابع لمؤسسة الميزانية الفيدرالية التابعة للدولة الفيدرالية ، وزارة الصحة الروسية. في سن الرابعة ، طورت الفتاة إفرازات تشبه الدورة الشهرية. كشفت الموجات فوق الصوتية عن كيس جرابي في المبيض الأيمن. تم إجراء عملية جراحية: إزالة كيس جرابي (استئصال المثانة). بعد العلاج الجراحي ، اختفت علامات البلوغ المبكر ، وكان معدل النمو طبيعيًا ، ولم يعد هناك تورم في الغدد الثديية.
بعد تحليل التاريخ الكامل للحياة (بما في ذلك البلوغ المبكر) ، وكذلك بيانات الأشعة السينية والتصوير المقطعي المحوسب ، كان يشتبه في متلازمة MRD. تم إرسال الفتاة إلى معهد طب الغدد الصماء لدى الأطفال ، حيث سبق لها العلاج.
بيانات من الدراسات المختبرية والوظيفية.
التحاليل السريرية للدم والبول: طبيعي.
اختبار الدم البيوكيميائي: الكالسيوم الكلي - 2.27 (القاعدة 2.02-2.6 مليمول / لتر) ، الكالسيوم المتأين - 1.1 مليمول / لتر (القاعدة 1.13-1.32 مليمول / لتر) ، الفوسفور غير العضوي - 3.66 مليمول / لتر (القاعدة 0.86-1.56 مليمول / لتر) / ل) ، مؤشرات أخرى ضمن النطاق الطبيعي.
التحليل البيوكيميائي للبول: طبيعي.
ملف الغدة الدرقية: TSH 3.2 µIU / لتر (عادي 0.32-5 µIU / لتر) ؛ إجمالي T4 65 نانومول / لتر (القاعدة 39-155 نانومول / لتر) ؛ T4 مجاني 21 نانومول / لتر (القاعدة 13-30 نانومول / لتر).
المظهر الهرموني: البرولاكتين 6.1 نانوغرام / مل (العادي 4-23 نانوغرام / مل) ؛ سوماتوميدين C 423ng / ml (القاعدة 95-473ng / ml) ؛ باراثورمون 16 نانوغرام / لتر (القاعدة 8-24 نانوغرام / مل) ؛ استراديول 24pg / مل (القاعدة 22-30pg / مل) ؛ FSH 6.8mU / لتر (معيار العمر 1.5-4.0mU / لتر) ؛ 1.75mU / ml (معيار العمر 0.5-4.6mU / l).
التصوير الومضاني للتكنيتيوم: هناك زيادة في تراكم العامل الدوائي في منطقة الثلث العلوي لعظم الفخذ الأيمن (102٪) ، في منطقة الثلث الأوسط من القصبة اليمنى (180٪) ، والكاحل الأيمن (106٪).
الأشعة السينية للجمجمة في الإسقاط الجانبي - لا علم الأمراض.
الموجات فوق الصوتية للغدة الدرقية: الحجم الكلي 6.6 سم 3 ؛ صدى متوسط. بنية الحمة هي بصيلات متوسعة غير متجانسة. الموجات فوق الصوتية للأعضاء الداخلية الأخرى بدون ميزات.
طرق البحث الآلية والمخبرية تستبعد أمراض الغدد الجار درقية والغدة النخامية والمشاش والغدد الكظرية. تم تأكيد التشخيص: "متلازمة MOD".
لم يتم بعد تطوير العلاج الموجه للسبب لخلل التنسج الليفي متعدد التوضعات.
يوصى به: تجنيب نظام تقويم العظام ، والتوقف عن الرقص ، وارتداء نعال لتقويم العظام ، وتجنب المجهود البدني والإصابات ، وتمارين العلاج الطبيعي لتقوية عضلات الأطراف السفلية ، ومنطقة أسفل الظهر ، والظهر ، والسباحة. تم وصف العلاج الدوائي: alfacalcidol 0.25 mcg 1 مرة في اليوم ، تحت سيطرة التحليل الكيميائي الحيوي ، عامل مضاد لهشاشة العظام (osteogenon 1 قرص مرتين في اليوم لمدة شهر واحد مرتين في السنة) ، منظم استقلاب الفوسفور والكالسيوم ( calcemin Advance 1 tablet مرتين في اليوم لمدة شهر واحد 4 مرات في السنة).
بعد مرور عام ، نتيجة لانتهاك النظام ، تلقت الفتاة كسرًا مغلقًا في الكتائب الوسطى للإصبع الرابع من القدم اليمنى في مركز الترامبولين. في الصور الشعاعية للقدم اليمنى في الإسقاطات المباشرة والمائلة ، يتم تصور كسر مرضي في الكتائب الوسطى للإصبع الرابع من القدم اليمنى ، وترقق الطبقة القشرية للأصابع الثانية والرابعة ، ويتم تمييز الحزم العظمية بشكل سيء. أنتجت ضمادة الجص الشلل لمدة 4 أسابيع. بعد هذه الفترة ، تُظهر الصور الشعاعية في الإسقاطات المباشرة والمائلة الانبساط بين الأجزاء ، والتوحيد المتأخر ، وهو أمر نموذجي لمتلازمة MRD.


الشكل 3 أ ، ب.الصور الشعاعية للقدم اليمنى: أ- قبل الشلل ، ب- بعد 4 أسابيع من الشلل
تم التخطيط للعلاج في المستشفى لضبط جرعة البايفوسفونيت. في الوقت الحالي ، لا يُنصح بالعلاج الجراحي ، لأنه مع بداية سن البلوغ ، من الممكن إبطاء أو حتى إيقاف التطور الإضافي لخلل التنسج العظمي. ينبغي النظر في إمكانية الجراحة للتكوينات الكيسية التي تهدد بشكل مباشر كسر مرضي.
1. في حالة الاشتباه في متلازمة MRD ، من الضروري إجراء دراسة شاملة عن سوابق الدم ، نظرًا لأن عدم وجود بيانات كاملة في السجلات الطبية ليس نادرًا.
2. معايير التشخيص: المظاهر الجلدية - وحمة لون القهوة مع الحليب ، التطور الجنسي المبكر ، خلل التنسج الليفي. نظرًا لأن الأشكال غير المكتملة والممحاة من متلازمة MRD أكثر شيوعًا ، يجب الاشتباه في وجود علامتين فقط.
3- يمكن أن تكون الدراسات المعملية (هرمونات المصل ، اختبار الدم البيوكيميائي) بدون تغيرات مرضية. قد تكون تشوهات الغدد الكظرية والغدة النخامية وعلامات زيادة النمو في الطول وتضخم الأطراف وما إلى ذلك غائبة أيضًا.
4. تشير نسبة FSH: LH> 2.5 (في هذه الحالة 3.9) إلى احتمال تكرار كيس المبيض والحاجة إلى المراقبة من قبل أخصائي الغدد الصماء.
5. الفحص الشعاعي الدوري ضروري للكشف عن تطور الأكياس الليفية.
قائمة الاختصارات.
ACTH هو هرمون موجه لقشر الكظر.
الفأس - شعر الإبط.
التصوير المقطعي المحوسب.
LH هو هرمون ملوتن.
العلاج بالتمرين - الثقافة الفيزيائية العلاجية.
أماه - الغدد الثديية.
متلازمة MRD - متلازمة ماكيون أولبرايت برايتسيف.
السارس هو عدوى فيروسية تنفسية حادة.
PTH - هرمون الغدة الجار درقية
ف - شعر العانة.
T4 - هرمون الغدة الدرقية.
TSH هو هرمون الغدة الدرقية.
الموجات فوق الصوتية - الموجات فوق الصوتية.
FP هو دواء دوائي.
FSH هو هرمون منشط للجريب.
cAMP هو أدينوزين أحادي الفوسفات.
المؤلفات.
1. Makazan N.V. دور اضطرابات إشارات ما بعد المستقبلات في تطوير المقاومة متعددة الهرمونات وفرط الوظائف اللاإرادي للغدد الصماء عند الأطفال. - ديس ... كان. عسل. العلوم. - M. ، 2017.
2. Uvarova E.V. الأساس المنطقي لاستخدام التجهيز النباتي على أساس Vitex agnus castusفي الفتيات المصابات بالبلوغ المبكر المرتبط بمتلازمة ماكيون أولبرايت برايتسيف // الصحة الإنجابية للأطفال والمراهقين. - 2017. - رقم 6. - ص 77–83.
3. Plaksina M.I.، Vitebskaya A.V. تقلب الأعراض في متلازمة ماكيون أولبرايت بريتسيف // طب الأطفال. - 2016. - رقم 6 (123).
4. متلازمة أولبرايت: الأسباب ، العلامات ، التشخيص ، كيفية العلاج // Syndrome.info: كل شيء عن المتلازمات. - http://sindrom.info/olbrajta/
متلازمة أولبرايت ، أو قصور جارات الدرق الكاذب ، هي مرض نادر جدًا محدد وراثيًا يتجلى في اضطرابات استقلاب الكالسيوم والفوسفور ، ونتيجة لذلك ، أمراض الجهاز الهيكلي. كقاعدة عامة ، تترافق الأعراض الرئيسية أيضًا مع تأخر في النمو - عقليًا وجسديًا. هذا المرض أكثر شيوعًا عند النساء.
المسببات
يعتقد العلماء أن متلازمة أولبرايت ترجع إلى خلايا الكلى والعظام السليمة لهرمونات الغدة الجار درقية. هذا بسبب خلل في المستقبلات. في الكلى ، يتم تعطيل تخليق cAMP (cyclic aminazine mophosphate) ، وهو رابط مساعد في عمليات التمثيل الغذائي المرتبطة بهرمون الغدة الجار درقية. يحدث هذا الضرر عندما تتلف جينات معينة.
يمكن أن يكون الانتهاك في أي جزء من سلسلة التمثيل الغذائي. في بعض المرضى ، يتضرر المستقبل نفسه ، وفي حالات أخرى ، تخضع بروتينات غشاء الخلية لتغييرات ، وفي حالات أخرى ، يرتبط المرض بعدم كفاية إنتاج cAMP. كل هذه الحالات مرتبطة بالشرطية الوراثية لعلم الأمراض.
طريقة تطور المرض
ترتبط متلازمة أولبرايت ارتباطًا وثيقًا بفسيولوجيا الغدد الجار درقية. عادة ، ينظمون مستوى الكالسيوم والفوسفور في الدم ، لكن في هذه الحالة المرضية لا يبالون بتقلبات هذه العناصر ، والتي يمكن أن تؤدي إلى فرط نشاط جارات الدرق الثانوي. ولكن حتى المستويات العالية من الباراهورمون لا تحفز التخلص السريع من الفوسفور و cAMP من الدم (حيث أن الكلى سليمة لتأثيراتها) وفي نفس الوقت تسبب تغيرات في بنية العظام.
كقاعدة عامة ، هناك زيادة تعويضية في الغدد الجار درقية ومسامية العظام وظهور التكوينات الكيسية فيها وتكلسات في الجلد والأنسجة الدهنية والعضلات والقلب والأوعية الدموية والملتحمة والقرنية.
عيادة

تتجلى متلازمة أولبرايت بنفس طريقة قصور الدريقات الحقيقي. يعاني المرضى من تشنجات ، والتي يمكن أن تحدث بمفردهم وبعد التعرض لعوامل مزعجة (التوتر العصبي ، تغيرات درجة الحرارة ، العمل البدني الشاق ، إلخ). يمكن أن تتقرح التكلسات الموجودة في الجلد والدهون تحت الجلد وتسبب عدم الراحة للمرضى. في هذه الحالة ، سيطلب المريض المساعدة ليس من أخصائي الغدد الصماء أو أخصائي الوراثة ، ولكن من الجراح أو طبيب الأمراض الجلدية.
عادة ما يكون هؤلاء الأشخاص قصيري القامة ولديهم وجه مستدير منتفخ وسمنة وأصابع قصيرة. تُلاحظ المفاصل الكاذبة حيث لا ينبغي أن تكون ، ولا توجد حركة في تلك الأماكن التي يُفترض فيها تشريحًا ، وهو انتهاك لتكوين المفاصل والعظام. تشمل الأعراض أيضًا القيء ووجود دم في البول وإعتام عدسة العين وتغيرات في مينا الأسنان.
بالإضافة إلى ذلك ، بسبب انخفاض مستويات الكالسيوم في الدم منذ الطفولة ، يعاني المرضى من انخفاض في الذكاء وتأخر النمو.
التشخيص

متلازمة أولبرايت هي مرض يصعب تشخيصه عند الرضيع ، ولكن في سن 5-10 سنوات ، تظهر مظاهر ملفتة للنظر لا تترك الطبيب في شك بشأن التشخيص. في الأطفال ، لوحظت تشوهات متعددة في الهيكل العظمي ، ينخفض مستوى الكالسيوم في الدم ويزداد مستوى الفوسفور وهرمون الغدة الدرقية.
لتأكيد التشخيص ، يمكن إجراء اختبار لكمية الفوسفات و cAMP في البول. للقيام بذلك ، يتم حقن المريض بـ 200 وحدة من هرمون الغدة الجار درقية عن طريق الوريد ثم يتم جمع جزء من البول بعد 4 ساعات. إذا لم يتم ملاحظة زيادة كبيرة ، فقد يشير ذلك إلى مقاومة أنسجة الكلى لهرمون الغدة الجار درقية.
هذه العلامات تكفي لوضع متلازمة أولبرايت. يمكن استكمال التشخيص بالتصوير الشعاعي. يجعل من الممكن رؤية تغييرات محددة في نظام الهيكل العظمي والأنسجة الرخوة للمريض.
بالإضافة إلى هرمون الغدة الجار درقية ، فإن الأشخاص الذين يعانون من هذا التشخيص يسجلون أيضًا مقاومة للهرمونات الأخرى ، لذلك يتم تشخيص النساء بالتشخيص التفريقي لمتلازمة شيريشيفسكي-تيرنر. هذان المرضان لهما تشابه خارجي ، وللتحقق من التشخيص ، يجب على الطبيب وصف تحليل الكروماتين الجنسي وإحالة الفحص النسائي والموجات فوق الصوتية لأعضاء الحوض.
العلاج والتشخيص

بمجرد أن يتم تشخيص متلازمة أولبرايت ، يجب بدء العلاج دون تأخير ، لأن التأخير يؤدي إلى تفاقم القصور العقلي. يتم وصف مكملات الكالسيوم للأطفال بجرعات يومية كافية للحفاظ على التركيزات الطبيعية في الدم. ثم يضاف إلى ذلك تناول فيتامين د بجرعة لا تتجاوز 100000 وحدة في اليوم.
لضبط العلاج ، من الضروري مراقبة مستوى الكالسيوم كل 7 أيام في الأسبوعين الأولين ، ثم مرة في الشهر حتى نهاية العلاج. عند الوصول إلى التركيز الأمثل للكالسيوم في الدم ، يمكن فحصه كل ثلاثة أشهر.
للقضاء على أعراض فرط نشاط جارات الدرق الثانوي ، يوصى باتباع نظام غذائي يحتوي على كمية منخفضة من الفوسفور. إذا تم الكشف عن نقص في الهرمونات الأخرى ، يتم إجراء العلاج البديل والأعراض.
إذا بدأ العلاج في الوقت المحدد وكان العلاج عقلانيًا ، فإن التكهن بالحياة يكون مناسبًا. ولكن قبل الحمل ، تحتاج النساء المصابات بهذا التشخيص إلى استشارة أخصائي علم الوراثة.
متلازمة أولبرايت هي مرض وراثي نادر ذو أصل غير معروف. إنها موروثة وتتميز بشكل أساسي بتلف الهيكل العظمي. غالبا ما توجد في الإناث. لكن في بعض الأحيان يمرض الأولاد أيضًا. يعاني معظم مرضى هذه المتلازمة من التخلف البدني والعقلي.
سمي المرض على اسم العالم الذي اكتشفه. يبدأ نظام الهيكل العظمي في الانهيار تحت تأثير استقلاب الكالسيوم والفوسفور. تفرز الغدة الجار درقية هرمون الغدة الجار درقية ؛ في الشخص المصاب بمتلازمة أولبرايت ، تضعف مقاومة الأنسجة لهرمون الغدة الجار درقية.
أعراض متلازمة بتلر أولبرايت
هناك ثلاث سمات رئيسيةالأمراض. في وجود اثنين من كل ثلاثة ، يمكن إجراء مثل هذا التشخيص.
- البلوغ المبكر جدا نتيجة لخلفية هرمونية عالية جدا. يمكن أن يبدأ الحيض عند الفتيات في الأشهر الأولى بعد الولادة. في الأولاد الذين يعانون من متلازمة أولبرايت ، نادرًا ما يلاحظ البلوغ المبكر. يحدث تغيير في الخلفية الهرمونية بسبب الأداء غير السليم للغدد الكظرية والغدة النخامية والغدة الدرقية. قد تظهر هذه الأعراض بشكل كامل أو غير مكتمل. مع الشكل الكامل للحيض ، فإنها تبدأ مبكرًا جدًا ، وعادة ما تكون وفيرة ومؤلمة. في مرحلة الطفولة ، تظهر هذه المتلازمة أيضًا خصائص جنسية ثانوية ، على سبيل المثال ، تورم الغدد الثديية. مع عدم اكتمال مظهر الحيض. في كثير من الأحيان ، مع متلازمة أولبرايت ، تصاب الفتيات بتكيسات على المبايض. في الأولاد الذين يعانون من سن البلوغ المبكر ، هناك زيادة في القضيب وشعر العانة.
- خلل التنسج العظمي الليفي. يشير هذا المرض إلى حدوث خلل في أنسجة العظام ، ويؤدي إلى انحناء وتشوه العظام. تتميز بعدم التناسق. العلامات الأكثر شيوعًا هي العرج وانحناء العمود الفقري. في أغلب الأحيان ، تخضع العظام الأنبوبية للتغييرات. يتباطأ نمو الهيكل العظمي ، وهو ملحوظ حتى عند الأطفال. يعاني العديد من ضحايا متلازمة ماكيون من نمو غير منتظم في عظام الجمجمة ، مما يؤدي إلى تشوه خارجي. مع حدوث تغيير في الوجه ، يكون مظهر العيون المنتفخة أمرًا ممكنًا. بعد بلوغ سن البلوغ ، تتباطأ تغيرات العظام ويقل تواتر الكسور.
- تغيرات الجلد. يتجلى في زيادة تصبغ. الجلد على الجذع والفخذين مغطى ببقع صفراء بنية ذات حواف غير متساوية. يمكن رؤية التصبغ في أسفل الظهر وأعلى الظهر والأرداف والصدر. تظهر البقع مباشرة بعد ولادة الطفل.

علاج متلازمة أولبرايت (ماكيون)
 المرضى الذين يعانون من متلازمة ماكون، يجب أن يكون دائمًا تحت إشراف متخصصين ضيقين (أخصائي أمراض النساء ، أخصائي الغدد الصماء ، أخصائي الرضوح ، طبيب العيون).
المرضى الذين يعانون من متلازمة ماكون، يجب أن يكون دائمًا تحت إشراف متخصصين ضيقين (أخصائي أمراض النساء ، أخصائي الغدد الصماء ، أخصائي الرضوح ، طبيب العيون).
العلاج الشامل مطلوب. نظرًا لأن المرض يتميز بطفرة جينية ، فمن الضروري أولاً وقبل كل شيء التخلص من أعراض مظاهره.
يشار إلى العلاج الهرموني الإلزاميلدعم نظام الغدد الصماء.
من الضروري عدم تفويت اللحظة التي تبدأ فيها عظام الوجه بالتشوه من أجل منع هذه التغييرات أثناء المرض.
في بعض الأحيان هو مطلوب تدخل جراحي. يتم وصفه في حالة وجود خطر الإصابة بفقدان السمع أو الرؤية بسبب التغيرات في بنية الجمجمة.
أثناء الألم ، يتم استخدام مسكنات الألم الخاصة التي تحتوي على كمية صغيرة من البايفوسفونيت.
من المهم جدًا القيام بإجراءات وقائية لتقوية العضلات.
لكل مريض يعاني من مرض أولبرايت ، يختار الطبيب علاجه الفردي الذي يجب اتباعه بدقة.
تحتاج إلى مراقبة باستمراركمية الكالسيوم في الجسم. في بداية العلاج ، يتم إجراء هذا الفحص مرة واحدة في الأسبوع ، ثم يتم تقليل تواتره إلى مرة واحدة في الشهر حتى يتم الانتهاء من العلاج الرئيسي.
من المهم جدًا أثناء العلاج اتباع نظام غذائي يتم فيه تقليل كمية المنتجات التي تحتوي على الفوسفور إلى الحد الأدنى.
وتجدر الإشارة إلى أن ، امرأة تعاني من هذه المتلازمة، عندما تقرر الحمل ، يجب عليك بالتأكيد استشارة اختصاصي وراثة جيد.
يصيب مرض أولبرايت (ماكيون) شخصًا واحدًا من بين كل مليون شخص ، ولم يتم حتى الآن اختراع علاج محدد لهذه المتلازمة ، لكن لا تفقد الأمل. الطب لا يقف ساكنا ، والأطباء سيساعدون في التغلب على اللحظات غير السارة وسيبقون المريض تحت الملاحظة طوال الوقت.
طب الأطفال Catad_tema - مقالات
قصور الدُّرَيْقات الكاذب (الحثل العظمي الوراثي لألبرايت): صعوبات في البحث التشخيصي التفاضلي. الملاحظة السريرية
إي. توزليان ، اختصاصي الغدد الصماء لدى الأطفال ، أخصائي الوراثة ، دكتوراه ، IV. شولياكوفا ، طبيب أعصاب ، دكتوراه ،
التقسيم الهيكلي المنفصل "معهد البحث العلمي السريري لطب الأطفال" SBEE HPE "الجامعة الوطنية للبحوث الطبية الروسية سميت باسم N.I. Pirogov "من وزارة الصحة في الاتحاد الروسي ، موسكو
الكلمات الدالة:
الأطفال ، قصور الغدد جارات الدرقية الكاذب ، حثل العظم الوراثي ألبرايت ، السمنة ، نقص كالسيوم الدم ، التشخيص ، مقاومة هرمون الغدة الجار درقية.
الكلمات الدالة:الأطفال ، قصور الغدد جارات الدرقية الكاذب ، حثل العظم الوراثي ألبرايت ، السمنة ، نقص كالسيوم الدم ، التشخيص ، مقاومة هرمون الغدة الجار درقية.
قصور جارات الدرقية الكاذب (الزائفة اليونانية - كاذبة + قصور جارات الدرقية ؛ مرادف: حثل عظمي وراثي لألبرايت ، متلازمة "الدجاج الجاوي") هو مرض وراثي نادر في نظام الهيكل العظمي الذي يحاكي قصور جارات الدرقية ويتميز بخلل في استقلاب الكالسيوم والفوسفور. غالبًا ما يكون مصحوبًا بتأخر في النمو العقلي والجسدي. تم وصف المرض لأول مرة من قبل أخصائية الغدد الصماء الأمريكية أولبرايت إف في عام 1942. معدل انتشار المرض هو 7.9 لكل مليون شخص.
البيانات الوراثية
قصور الدريقات الكاذب (PHP) هو مرض غير متجانس وراثيا. البيانات المتعلقة بنوع الانتقال الوراثي متناقضة: كلا النوعين المتنحيين الوراثي المتنحي المرتبط بالكروموسوم X والمسيطر عليه. في معظم الحالات ، يرتبط تطور الحثل العظمي الوراثي لألبرايت بطفرات في الموضع 20q13 من جين GNAS1 الموجود على الكروموسوم 20 (باتن وآخرون ، 1990) ، والذي يشفر بروتين Gs-alpha المرتبط بهرمون الغدة الجار درقية (PTH) مستقبل. تم العثور على نمط ظاهري مشابه أيضًا في المرضى الذين يعانون من الحذف الخلالي للذراع الطويل للكروموسوم 2 للموضع 2q37.
طريقة تطور المرض
يعتمد التسبب في قصور جارات الدرقية الكاذب على المقاومة المحددة وراثيًا للكلى والهيكل العظمي لعمل الباراثورمون نتيجة لخلل في معقد "مستقبلات خلوية محددة - هرمون الغدة الجار درقية - إنزيم الأدينيلات الحلقي" ، مما يعطل تكوين الحلقة 3 "- ، 5 "-أدينوسين أحادي الفوسفات (cAMP) في الكلى ، وهو وسيط داخل الخلايا لعمل هرمون الغدة الجار درقية على عمليات التمثيل الغذائي. قصور الدريقات الكاذب وراثي مرض غير متجانس.في بعض المرضى ، يكون المستقبل الخلوي نفسه معيبًا ، والذي يرتبط بهرمون الغدة الجار درقية (النوع 1A قصور جارات الدرقية الكاذب) ، بينما يعاني البعض الآخر من خلل في البروتين المرتبط بالنيوكليوتيدات المترجمة في الطبقة الدهنية ثنائية الغشاء الخلوي ويربط المستقبل وظيفيًا بحلقة الغدد الجار درقية (النوع) 1B قصور الدريقات الكاذب). يعاني بعض المرضى من نقص إنزيمي في إنزيم adenylate cyclase نفسه (قصور جارات الدرقية الكاذب من النوع 2). يؤدي نقص cAMP ، الناتج عن هذه العيوب ، إلى انتهاك تخليق بروتينات معينة تحدد التأثير البيولوجي لهرمون الغدة الجار درقية. وبالتالي ، يتم فقدان حساسية الأعضاء المستهدفة لهرمون الغدة الجار درقية.
الخصائص السريرية
حاليًا ، يتم تمييز 4 أشكال سريرية من علم الأمراض: الأنواع 1A ، 1B ، 1C و 2. تسمح المعرفة بخصائصها السريرية والكيميائية الحيوية وبيانات البحث الجيني بالتشخيص التفريقي ضمن الشكل التصنيفي نفسه.
العلامات العامة التي تسمح بالشك في المرض هي التطور البدني غير المتناسب ، وقصر القامة (حتى القزامة) بسبب تقصير الأطراف السفلية (الصورة 1) ، العضدي (الصورة 2) ، الوجه المستدير "على شكل القمر" (الصورة 3). في بعض الأحيان يكون هناك عوارض وتضخم للأسنان.
الصورة 1.
ظهور طفل مصاب بالحثل العظمي أولبرايت
(سمات النمط الظاهري ، قصر القامة بسبب تقصير الأطراف السفلية)
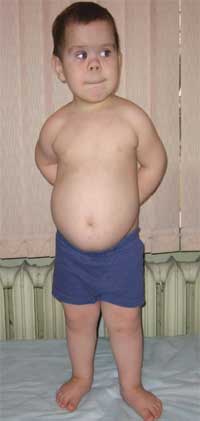
الصورة 2.
ملامح الهيكل العظمي للمريض
مع Albright osteodystrophy
(اصابع قصيرة - اصابع قصيرة)
صورة 3.
ملامح النمط الظاهري للطفل
مع Albright osteodystrophy
(وجه "قمر" مستدير)

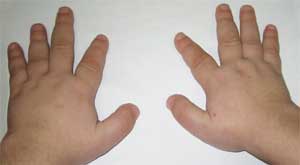
يعتبر التقصير الحاد في عظام المشط الأول والثالث والخامس (خاصة الثالثة والرابعة) علامة مرضية ، ونتيجة لذلك تكون الأصابع الثانية على اليدين والقدمين أطول من الأصابع الأخرى ، وعندما تكون اليد كذلك مشدودة في قبضة ، لا توجد انتفاخات في منطقة المفاصل السنعية السنية الرابعة والخامسة - ما يسمى بالانتفاخ العضدي. الكتائب العريضة القصيرة ، سماكة قبو الجمجمة ونزع المعادن من العظام (هشاشة العظام) ، السمنة تم الكشف عنها أيضًا.
تم العثور على التخلف العقلي (غالبًا ما يكون متوسط الشدة) في حوالي 20 ٪ من المرضى. وفقًا لبعض المؤلفين ، تحدث قلة القلة في 70 ٪ من حالات نقص كلس الدم وفي 30 ٪ من الحالات المصابة بسرطان الدم. تتباطأ العمليات العقلية عند المرضى. في الحالة العصبية ، غالبًا ما يتم ملاحظة الإحراج الحركي والتفاعلات العصبية: مخاوف ، قلق ، قلق ، قلة النوم ، زيادة ردود الفعل ، تشنجات كزازية وتسببها نقص كالسيوم الدم ، نوبات متشنجة في بعض الأحيان. يتم وصف أعراض الاعتلال العضلي أيضًا: إرهاق عضلي ، ضعف عضلي. غالبًا ما تُلاحظ اضطرابات خارج السبيل الهرمي: فرط الحركة ، كنع ، نصف تشنج في الوجه ، باركنسون ، في بعض الحالات نوبات صرع ، أعراض مخيخية: ترنح ، ضعف التنسيق.
في كثير من الأحيان ، يتم تحديد تكلسات الأنسجة الرخوة والتكلسات تحت الجلد (الصدر والبطن والأوتار العظمية) ، مع الفحص النسيجي منها - ورم عظمي(Izraeli وآخرون ، 1992) ، الدماغ (العقد القاعدية). من المهم ملاحظة أن التكلسات قد تكون موجودة بالفعل عند الولادة. بسبب نقص كالسيوم الدم ، عادةً ما يتطور إعتام عدسة العين وتحدث عيوب في مينا الأسنان.
التهاب الغدة الدرقية من النوع 1 أ
لديه وضع وراثي جسمي سائد في الميراث. يقع جين قصور جارات الدرقية الكاذب من النوع 1A ، GNAS1 ، على الذراع الطويلة للكروموسوم 20 ، في موضع 20q13.2. يرتبط تطور المرض بنقص بروتين ربط الجوانين بالنيوكليوتيدات (بروتين Gs). في الوقت نفسه ، فإن هرمون PTH ، المرتبط بمستقبلات الأنسجة المستهدفة ، غير قادر على تنشيط أحادي فوسفات الأدينوزين الدوري (cAMP) ويسبب استجابة الأنسجة. على الأرجح ، هناك آلية مماثلة تكمن وراء تطور عدم حساسية أنسجة الأعضاء الأخرى والغدد الصماء (قصور الغدة الدرقية ، الغدد التناسلية ، الغدة النخامية ، داء السكري ، وكذلك انخفاض استجابة الكبد لإدارة الجلوكاجون) التي لوحظت في النوع 1A قصور جارات الدرقية الكاذب. مع هذا النوع من الأمراض ، لا يوجد زيادة في إفراز cAMP في البول ، وهو ما يميز القاعدة ، استجابةً للإعطاء الخارجي لـ PTH. يتم تشخيص المرض في كثير من الأحيان في سن 5-10 سنوات. يعاني المرضى من قصر القامة ، وعنق قصير ، ووجه مستدير ، وتقصير في عظام المشط والمشط (في كثير من الأحيان تقصير الأصابع الوريدية وغالبًا ما تكون الأصابع الثانية) - ما يسمى بالمثل العضدي. لوحظ تكلس الأنسجة الرخوة ، تكلسات تحت الجلد ، والتي يمكن اكتشافها بالفعل عند الولادة ؛ غالبًا ما يكون هناك إصابة متزامنة بالغدد الصماء الأخرى: الغدة الدرقية (قصور الوظيفة) ، الغدد التناسلية ، البنكرياس (داء السكري). غالبًا ما يحدث إعتام عدسة العين وخلل في مينا الأسنان بسبب نقص كالسيوم الدم. كاختبار تشخيصي تفاضلي للفرق بين PPH من النوع 1A وقصور جارات الدرقية: عدم وجود تأثير سريري من الإعطاء بالحقن لـ PTH في شكل زيادة في مستوى الكالسيوم في الدم وزيادة في إفراز الكلى للفوسفور في البول (تأثير الفوسفاتوريك).
يكشف الفحص البيوكيميائي عن نقص كالسيوم الدم ، فرط فوسفات الدم ، زيادة في مستوى هرمون الغدة الجار درقية في الدم ، ونقص فوسفات الدم. ينخفض مستوى البروتين Gs في الدم. يكشف الفحص بالأشعة السينية لنظام الهيكل العظمي عن قصر عظام المشط والمشط ، ونزع معادن معمم ، وسماكة عظام قبو الجمجمة.
PSEUDOHYPOPARATHYROISIS من النوع 1 ب
له نوع وراثي سائد من الميراث ، ومع ذلك ، لا يتم استبعاد نوع وراثي مهيمن مرتبط بـ X. من الضروري أن تضع في اعتبارك الاختراق غير الكامل الملحوظ أحيانًا لجين المرض وإمكانية النقل الخفي لعلم الأمراض. لذلك ، فمن المستحسن السريرية (الكشف عن المسار السريري للمرض) والفحص البيوكيميائي (تحديد مستويات الكالسيوم والفوسفور والدم PTH) للحوامل المزعومة للمرض. يحدث نوع HPT 1B بسبب نقص مستقبلات الأنسجة لهرمون الغدة الجار درقية في الأعضاء المستهدفة ومقاومة محدودة لهرمون الغدة الجار درقية. الصورة السريرية مشابهة للعيادة من النوع 1A ، ولكن لا يوجد ضرر للغدد الصماء الأخرى ، كما أن الحثل العظمي أقل شيوعًا.
في المرضى ، لا يوجد تفاعل كلوي للإعطاء الخارجي لهرمون الغدة الجار درقية على شكل زيادة في إفراز الأدينوزين أحادي الفوسفات الدوري في البول ، ومع ذلك ، على عكس النوع 1A ، فإن مستوى بروتين Gs في الدم طبيعي. تتأثر النساء أكثر من الرجال ، لكن شدة المرض يمكن أن تكون هي نفسها لدى كل من الرجال والنساء.
التهاب الغدة الدرقية من النوع 1 ج
يتعرف بعض المؤلفين على قصور جارات الدرقية الكاذب (PPHP) الذي وصفه أولبرايت إف في عام 1952. يتميز بالصورة السريرية المميزة لـ PAH ، لكن مستويات الكالسيوم والفوسفور في الدم والبول تظل ضمن المعدل الطبيعي. تظل مؤشرات PTH و Gs-protein في الدم أيضًا عند المستوى الطبيعي. يعاني بعض مرضى PHP من النوع 1C من عمليات حذف من جديدعلى الكروموسوم 2. من الممكن أن يكون هذا النوع من المرض هو نوع فرعي من HPP type 1A.
PSEUDOHYPOPARATHYROISIS من النوع 2
سريريًا مشابه لأنواع أخرى من المرض ، ولكن لديه وضع وراثي وراثي متنحي. لا يتم استبعاد وجود أشكال جسمية سائدة من علم الأمراض. يرتبط التسبب في التطور بالمقاومة داخل الخلايا لـ cAMP. في هذه الحالة ، يرتبط PTH بالمستقبلات ويسبب استجابة خلوية طبيعية لـ PTH في شكل زيادة في إفراز cAMP. ومع ذلك ، فإن الحساسية داخل الخلايا تجاه cAMP لا تسمح بالإدراك الكامل لعمل PTH. في الوقت نفسه ، يتم الحفاظ على التفاعل الطبيعي للكلى للإعطاء الخارجي لهرمون الغدة الدرقية في شكل زيادة في إفراز الأدينوزين أحادي الفوسفات الدوري في البول. تم اقتراح أن النوع 2 HWP قد يترافق مع نقص فيتامين (د).
وبالتالي ، فإن الأنواع المحددة من PHP تتميز سريريًا بانخفاض حساسية الأعضاء المستهدفة تجاه الهرمون الجارعي المغذي ، ولكنها تختلف في الآليات المسببة للأمراض لتشكيل عدم حساسية الأنسجة.
التشخيص
يمكن أن تكون طبيعة إفراز cAMP الكلوي استجابةً لإعطاء PTH بمثابة اختبار تشخيصي تفاضلي مختبري: لوحظ زيادة إفراز cAMP في النوع 2 وغيابه في النوع 1. يتم تأكيد التشخيص من خلال الكشف عن انخفاض مستوى النوكليوتيدات الغوانين. بروتين ملزم (Gs- بروتين) في الدم (بمعدل 1.5-2 مرة) مقارنة مع القاعدة. عادة ما يرتبط نقص كالسيوم الدم بفرط فوسفات الدم ونقص فوسفات الدم. مستوى PTH مرتفع ؛ مع النوع 1C ، يكون مستوى الهرمون الجار درقي طبيعيًا ، مما أدى إلى ظهور اسم "قصور جارات الدرق الكاذب". يكشف الفحص بالأشعة السينية للجهاز الهيكلي عن قصر في عظام المشط والمشط ، وغالبًا ما يتم إزالة المعادن بشكل عام (هشاشة العظام) ، وسماكة عظام قبو الجمجمة. يُظهر الرسم الجلدي إزاحة الراحي المحوري ثلاثي نصف القطر.
معايير التشخيص:
- النمو المنخفض؛
- وجه مستدير؛
- تأخر النمو العصبي النفسي.
- تشوهات هيكلية
- انخفاض الكالسيوم في الدم
- ارتفاع مستويات هرمون الغدة الجار درقية في الدم.
- انخفض إفراز البول للفوسفات و cAMP.
العلاج والوقاية
يتكون علاج نقص كالسيوم الدم من تعيين مكملات الكالسيوم بجرعات كافية للحفاظ على التركيز الطبيعي للكالسيوم في الدم. يعتبر العلاج بفيتامين (د) ذا أهمية كبيرة. حاليًا ، يتم استخدام المستقلبات النشطة لفيتامين د - أوكسيديفيت ، 1-ألفا-د 3 ، كالسيترين ، إلخ بجرعة 1-2 ميكروغرام / يوم بنتيجة إيجابية (زيادة الكالسيوم في الدم ، انخفاض مظاهر المتلازمة المتشنجة). Tachystin (0.5-1.5 مجم / يوم) فعال أيضًا. يزيد هذا الدواء من امتصاص الكالسيوم في الأمعاء وبالتالي يزيد من مستوى الكالسيوم في الدم. يستخدم العلاج المضاد للاختلاج كعلاج مساعد. العلاج ليس له تأثير ملحوظ على التطور الفكري ، ولكن إلى جانب انخفاض أعراض متلازمة الاختلاج ، لوحظ تراجع المظاهر العصبية (الاضطرابات تحت القشرية ، فرط الحركة المشيمية ، الكُنْع ، إلخ). من أجل تجنب جرعة زائدة من مستحضرات فيتامين د ، من الضروري التحكم في تركيز الكالسيوم في الدم كل 3-7 أيام خلال الأسبوعين الأولين من العلاج وكل شهر لمدة 2-3 أشهر القادمة. عند الوصول إلى تركيز ثابت من الكالسيوم في الدم ، يكفي فحصه مرة كل شهرين إلى ثلاثة أشهر. يساعد النظام الغذائي المقيّد بالفوسفور على تطبيع تركيز الفوسفور والكالسيوم في الدم والقضاء على أعراض فرط نشاط جارات الدرق الثانوي. في حالة قصور الغدد الصماء الأخرى ، يتم إجراء العلاج البديل بالهرمونات المناسبة.
العلاج بهرمون الغدة الدرقية غير فعال. للتخفيف من النوبات المتشنجة ، يتم إعطاء محلول 10 ٪ من كلوريد الكالسيوم أو غلوكونات الكالسيوم عن طريق الوريد ؛ الداخل - محلول 5-10 ٪ من كلوريد الكالسيوم ، ملعقة كبيرة 3-4 مرات في اليوم: غلوكونات الكالسيوم ، لاكتات الكالسيوم - ما يصل إلى 10 غرام في اليوم.
تنبؤ بالمناخمدى الحياة يتحدد من خلال شدة متلازمة التشنج.
منعيعتمد المرض على بيانات الاستشارة الوراثية الطبية.
استشارة طبية وراثية
في الاستشارة الوراثية الطبية ، يجب على المرء أن ينطلق من نوع وراثي سائد من الوراثة وخطر مرتفع (50٪) لتكرار المرض في الأسرة ذات الأشكال الموروثة. من أجل تحديد طبيعة نوع الميراث ، من الضروري إجراء فحص شامل للوالدين ، حيث يمكن أن تظهر المتلازمة مع الحد الأدنى من الأعراض السريرية. حاليًا ، تم تطوير التشخيص الجيني الجزيئي للمرض ويتم تحسينه عن طريق تحديد الطفرات في جين GNAS1 على الكروموسوم 20. طرق التشخيص قبل الولادة للمرض ككل وأنواعه الفردية يجري تطويرها.
الملاحظة السريريةتم قبول Boy G. ، البالغ من العمر 14.5 عامًا (الصورة 4) ، في معهد الأبحاث السريرية لطب الأطفال بتشخيص مرض تنكسي في الجهاز العصبي؟ استسقاء الرأس الخارجي الخلقي. أعراض الصرع. متلازمة وراثية؟ مرض التخزين؟ اعتلال الدماغ الأيضي. قصور الغدة الدرقية تحت الإكلينيكي. مكانة قصيرة من التكوين المختلط ؛ الضعف الادراكي.
شكاويعند الدخول في صداع انتيابي شديد ، موضعي في المنطقة الأمامية ويصاحبه قيء ، مما يؤدي إلى الشعور بالراحة ، وانخفاض الذاكرة والأداء المدرسي ، والنوبات المتشنجة ، والتي يحدث خلالها ارتعاش في اليد اليمنى.
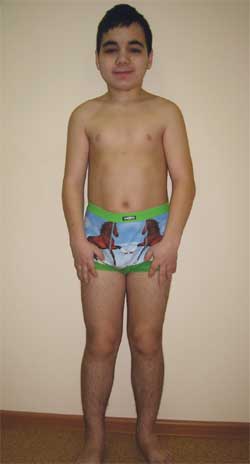
صورة 4.
الطفل ج. ، 14.5 سنة ، مصاب بالحثل العظمي أولبرايت
(ملامح النمط الظاهري ، قصر القامة ، تقصير الأطراف ، عضلي)
تاريخ العائلة:الوالدان أرمنيان بالجنسية ، لا تربطهما صلة قرابة بالدم وليس لديهم مخاطر مهنية. لم تكن هناك حالات مرض عقلي أو صرع أو تأخر في النمو في النسب. أشقاء ، أخت ، 17 سنة - من كلام - بصحة جيدة.
تاريخ الحياة والمرض:ولد من الحمل الثاني الذي مضى بدون ملامح ، ولادة ثانية ، في الوقت المناسب ، فسيولوجي ، وزن عند الولادة - 3100 غرام ، طول - 51 سم ، صرخ على الفور ، درجة أبغار - 7/9 نقاط. تدهور في اليوم الثالث - تشنجات حديثي الولادة ، توقفت في مستشفى الولادة. فترة ما بعد الولادة المبكرة - بدون ميزات. كان هناك تأخير بسيط في وتيرة التطور الحركي في السنة الأولى من العمر ، والمشي المستقل من سنة واحدة و 3 أشهر. في هذا الصدد ، لاحظه طبيب أعصاب بتشخيص الضرر العضوي للجهاز العصبي المركزي ؛ استسقاء الرأس الخلقي. تشنجات حديثي الولادة التشنجات الحموية في التاريخ.
تلقى دياكارب ، فينليبسين. ظهور النوبات لأول مرة منذ عام واحد و 11 شهرًا. - غير متماثل ، مقوي على شكل شد في الذراع اليمنى والساق ، مع ثبات العينين ، حتى دقيقتين ، دون فقدان للوعي ، متكرر حتى 10 نوبات في اليوم. تلقى ديباكين بشكل غير منتظم. على خلفية الإلغاء الذاتي - حالة منشط واحد. في سن الثانية ، تم إجراء فحص بالأشعة المقطعية للدماغ في مكان الإقامة ، حيث تم الكشف عن بؤر واحدة لإزالة الميالين في الفص القذالي.
باستشارة جراح أعصاب ، يوصى بالعلاج المحافظ. من سن 3 سنوات ، هناك تأخير في التطور النفسي ، يوصى بإشراف طبيب نفسي.
من سن 4-5 سنوات ، بدأ الآباء يلاحظون تشوه وقصر أصابع القدم واليدين ، وخاصة الأصابع II-IV بشكل متماثل في اليدين والقدمين ، وانخفاض في معدلات النمو. في سن الثامنة ، استنتاج معالج النطق هو اضطراب عام في النطق من المستوى الثاني أو الثالث ، يوصى بالتدريب في مدرسة متخصصة. في نفس العمر فحص من قبل اختصاصي الوراثة في محل الإقامة ، الاستنتاج: مرض التمثيل الغذائي الوراثي؟ تمت التوصية بدراسة الأحماض الأمينية في الدم ، ولم يتم الكشف عن أي تغييرات ؛ الاستنتاج النهائي: لم يتم الكشف عن بيانات مرض التمثيل الغذائي الوراثي. نقص التنسج الغضروفي. العلاج الموصى به من قبل طبيب أعصاب وأخصائي غدد صماء.
الطاولة.
لمحة عن النمو العقلي للطفل G. ، 14.5 سنة (معدل الذكاء = 68)

في سن الثامنة ، تمت استشارته من قبل أخصائي الغدد الصماء حول النمو وتأخر النمو. عند فحص اليدين بالأشعة السينية ، لوحظت السمات التالية: تم تقصير وتثخين عظام الكتائب الوسطى والكتائب الرئيسية وعظام المشط ؛ كان تشخيص اختصاصي الأشعة هو الودانة.
يتم فحصه بشكل متكرر في مكان الإقامة في مستشفى الأمراض العصبية. في سن 12 ، ظهرت نوبات تشنجية دون فقدان الوعي مع ارتعاش في اليد اليمنى ، والتي هي ذات طبيعة متسلسلة ، تم وصف العلاج المضاد للاختلاج (Depakine) ، وانخفض تواتر النوبات بشكل ملحوظ. في سن 13 ، تم إجراء التصوير بالرنين المغناطيسي للدماغ مع التباين - تغييرات متناظرة في قاعدة الفص الصدغي على مستوى النوى في شكل زيادة في إشارة MR ، وهو أمر نموذجي للسامة (المنغنيز) أو اعتلال الدماغ الأيضي (النحاس والحديد).
أعيد الفحص في سن 13 سنة 3 شهور. طبيب الغدد الصماء ، في دراسة ملف الغدة الدرقية ، تم الكشف عن زيادة في هرمون الغدة الدرقية (TSH) ، وتم تشخيص قصور الغدة الدرقية تحت الإكلينيكي ، وتم وصف هرمون الغدة الدرقية L.
عند تحليل بطاقة العيادة الخارجية للطفل والوثائق في مكان الإقامة ، تم إجراء دراسة الكالسيوم والفوسفور مرة واحدة ، في سن 1.5 سنة ، لوحظ نقص كلس الدم ، ولكن لم يتم إجراء فحص إضافي بهذه المناسبة. نظرًا لعدم اليقين في التشخيص في مكان الإقامة ، أرسل عالم الوراثة الطفل إلى موسكو ، إلى معهد البحث العلمي السريري لطب الأطفال ، من أجل توضيح التشخيص.
بيانات البحث الموضوعي:
الارتفاع - 143 سم ، الوزن - 43 كجم.
النمو البدني منخفض للغاية ومتناغم ، واللياقة البدنية غير متناسبة بسبب تقصير الأطراف. نمو Sds يتوافق مع -2.8 انحراف عن القاعدة (عادي -2 + 2).
ميزات النمط الظاهري: وجه مستدير ، وعنق قصير ، وشق مضاد للمغولويد للشقوق الجفنية ، وجسر أنف عريض ، وجبهة عالية ، وشبه عضلي ، وتقصير عظام المشط الرابع والخامس والمشط (الصورة 5). على الأعضاء الداخلية - بدون ميزات. التطور الجنسي - مرحلة تانر الثالث والرابع (التي تتوافق مع العمر).
بيانات من الدراسات المختبرية والوظيفية:
التحليل السريري للدم والبول هو القاعدة.
اختبار الدم البيوكيميائي: الكالسيوم الكلي - 1.39 (عادي 2.02-2.6 مليمول / لتر) ، الكالسيوم المتأين - 0.61 (طبيعي 1.13-1.32 مليمول / لتر) ، الفوسفور غير العضوي - 3.66 (القاعدة 0.86-1.56 مليمول / لتر) ، المؤشرات الأخرى ضمن المعدل الطبيعي.
التحليل الكيميائي الحيوي للبول: يتم تقليل إفراز الكلى للفوسفات - 11.5 مليمول / لتر (القاعدة 19-32 مليمول / لتر).
ملف الغدة الدرقية: TSH - 11.75 (القاعدة 0.4-4.0 μIU / ml) ، T4 مجاني - 0.49 (القاعدة 1.0-1.8 نانوغرام / ديسيلتر).
هرمون الغدة الجار درقية - 499 (طبيعي 12-65 بيكوغرام / مل) ، هرمون النمو - 7 نانوغرام / مل (طبيعي 7-10 نانوغرام / مل) ، سوماتوميدين- C - 250 نانوغرام / مل (طبيعي 88-360 نانوغرام / مل).
الموجات فوق الصوتية للأعضاء الداخلية - بدون ميزات.
ECG - هجرة منظم ضربات القلب فوق البطيني على خلفية معدل ضربات القلب المنتظم من 71-80 نبضة / دقيقة. حصار غير كامل للساق اليمنى من صرة له. انتهاك عملية عودة الاستقطاب في عضلة القلب للجدار الخلفي للبطين الأيسر (انخفاض في z.T III ، aVF).
R- الرسم البياني للعمود الفقري - الجنف الأيمن للعمود الفقري الصدري من الدرجة الأولى ، هشاشة العظام الشديدة.
R- رسم بياني لليدين مع إمساك الساعدين - تقصير وتوسيع الكتائب الطرفية والوسطى. عمر العظام - 13.5 - 14 سنة.
لم يتم تسجيل أنماط نشاط الصرع EEG.
التصوير بالرنين المغناطيسي للدماغ - صورة التصوير بالرنين المغناطيسي لبؤر متعددة تحت القشرية لزيادة إشارة MR في الفص الجبهي ، استسقاء الرأس الخارجي مع ضمور مادة الدماغ.
MSCT للدماغ - مناطق متناظرة من تكلس النوى العدسية. منتشر في مناطق شديدة الكثافة في المهاد ، نوى مذنبة مع منطقة تكلس على اليمين. تكلسات متعددة منقط للأنسجة الرخوة غلافية للجمجمة.
مخطط سمعي - بدون علم الأمراض.
يجري تشخيص الحمض النووي في جين GNAS1.
نصيحة إختصاصية:
أخصائي الغدد الصماء - الحثل العظمي الوراثي لألبرايت من النوع 1A (قصور الغدد جارات الدرقية الكاذب). قصور الغدة الدرقية الأولي ، تعويض طبي غير كامل.
طبيب عيون - إعتام عدسة العين الثانوي الكامل. العلاج الجراحي الموصى به.
عالم نفسي - إعاقات معرفية (يتم عرض الملف النفسي للطفل في الجدول).
مع الأخذ في الاعتبار النمط الظاهري للطفل ، وبيانات التاريخ ، ونتائج الدراسات الإضافية (نقص كالسيوم الدم ، فرط فوسفات الدم ، نقص الفوسفات ، زيادة هرمون الغدة الجار درقية في الدم) ، التكلسات في مادة الدماغ ، وجود إعتام عدسة العين ، قصور الغدة الدرقية) ، كان التشخيص مصنوع: حثل عظمي وراثي من نوع أولبرايت 1A (قصور جارات الدرق الكاذب). يوصى بإجراء تشخيص الحمض النووي - البحث عن الطفرات في جين GNAS1.
علاج او معاملة:ينصح الطفل بتناول euthyrox بجرعة 100 ميكروغرام / يوم ؛ المستقلب النشط لفيتامين D - alpha-D3 ("Teva") بجرعة 2 ميكروغرام / يوم ؛ الكالسيوم ("ساندوز") 2000 ملغ / يوم ؛ التناول المستمر للعلاج المضاد للاختلاج - فينليبسين 800 ملغ / يوم تحت إشراف طبيب أعصاب - صرع ؛ فصول مع أخصائي أمراض عيوب النطق وطبيب نفساني ؛ العلاج الموجه للطاقة (Elkar and coenzyme Q10 في الجرعات العمرية). التحكم في مؤشرات استقلاب الفوسفور والكالسيوم ومستويات هرمون الغدة الجار درقية.
في هذا الطريق،توضح الملاحظة السريرية المقدمة مدى تعقيد البحث التشخيصي التفاضلي ، وأهمية الدراسة في الوقت المناسب للمعلمات البيوكيميائية البسيطة (في حالة الصرع ، يعد الفحص المتكرر لمؤشرات استقلاب الفوسفور والكالسيوم إلزاميًا) ، ونتائج التشخيص المتأخر لمرض محدد وراثيًا ، الحاجة إلى دمج العلامات الفردية في النمط الظاهري العام لحالة مرضية معينة من أجل التشخيص المستهدف في الوقت المناسب لأشكال معينة من الأمراض الوراثية. يعد التشخيص في الوقت المناسب وتوضيح نشأة كل متلازمة أمرًا مهمًا بشكل خاص ، لأنها تتيح لك العثور على أفضل نهج لعلاج هذه الحالات ، والوقاية من المضاعفات المحتملة (حتى إعاقة الطفل) ؛ الوقاية من تكرار الإصابة بأمراض وراثية في الأسر المصابة (استشارة طبية وراثية). هذا يفرض الحاجة إلى الأطباء من مختلف التخصصات للتنقل بوضوح في تدفق علم الأمراض الوراثي. الببليوغرافيا قيد المراجعة.